All published articles of this journal are available on ScienceDirect.

De-Novo Protein Design Strategies for Targeting Rotavirus Proteins: Evolutionary Conservation and Therapeutic Implications
Abstract
Introduction
Rotavirus (RV) is a leading cause of pediatric gastroenteritis, with existing vaccines showing reduced efficacy due to strain diversity and limited immunogenicity. This study aimed to design de novo proteins targeting highly conserved RV proteins to develop potential therapeutic inhibitors.
Methods
Protein sequences from nine RV strains were retrieved from UniProtKB and aligned using T-Coffee to identify conserved regions. Structural modeling was performed with OmegaFold and SWISS-MODEL, and domain analysis was carried out via InterPro. Protein–protein docking with ClusPro and HDOCK identified interactions between viral proteins (VP4, VP7, RNA-dependent RNA polymerase) and host proteins (integrins and VP2). De novo proteins were computationally designed using residue-frequency constraints and screened for stability (DeepStabP), toxicity (CSM-Toxin), and docking affinity to RV targets.
Results
VP4, VP7, and RNA polymerase were the most conserved proteins across RV strains (77–85%). Domain-specific interactions with integrins and VP2 were identified. Designed proteins exhibited high binding affinities, notably with VP4 membrane interaction domains, VP7 domain 1, and RNA polymerase C-terminal regions. Over 96% of the designed sequences were predicted to be non-toxic, and most showed favorable thermal stability. Docking revealed conserved interaction sites across strains, suggesting broad-spectrum inhibitory potential.
Discussion
These findings demonstrate that de-novo-designed proteins can selectively target conserved rotavirus domains, potentially overcoming strain variability and vaccine limitations. While computational results support structural stability and non-toxicity, experimental validation is essential. Limitations include reliance on in-silico predictions and the absence of in-vivo confirmation.
Conclusion
This study highlights designing de novo proteins as a promising approach for developing novel antivirals against rotavirus, warranting further experimental and clinical investigations.
1. INTRODUCTION
Rotavirus (RV) is a member of the Sedoreoviridae family and the Orthornavirae kingdom [1]. It causes acute to severe gastroenteritis in infants and young children. It is a fatal disease causing high death rates in low-income countries, and the cases of diarrhea that have been reported every year in children and infants worldwide are greater than 125 million [2, 3]. The death rate recorded from diarrhea in 2019 was approximately 19.11%; however, the annual mortality rate from RV infection ranges between 20% and 40% [3, 4].
RV has A, B, C, D, E, F, G, and H strains [5]. It is a double-stranded RNA virus that has six structural (VP1, VP2, VP3, VP4, VP6, and VP7) and six non-structural (NSP1-NSP6) proteins, which form the three-layer capsid shell and are necessary for the replication cycle of the virus within the host [1]. RV enters the environment and infects individuals through the fecal or oral route. It is excreted in large quantities by infected humans, who are most likely to transmit the virus during the early stages of illness and for up to three days after recovery [6]. Moreover, high temperature, vomiting, gastroenteritis, and dehydration are some symptoms of RV [7].
The virus can enter the body through interactions between the host cell surface receptors and the virus. The entry of RVs into a cell involves proteolysis, attachment to cell-surface receptors, binding to the double-layer particles, disintegration of the DLP particle, and cessation within the cell cytoplasm [8]. Furthermore, the penetration of RV depends on several proteins, including the lectin-binding protein VP4, which is associated with P-type, also known as the protease-sensitive protein required for viral spread. Proteolytic cleavage of VP4 induces a conformational change that disrupts the host cell membrane, facilitating viral penetration [9]. Moreover, the glycoprotein VP7, also known as the G type, plays a key role in viral attachment and entry [10]. VP8 is also responsible for the initial attachment of a virus to the host cell [9].
The RotaTeq and Rotarix vaccines are reported to be effective against RV and are approved by the WHO (World Health Organization). Rotarix is a monovalent vaccination, whereas RotaTeq is a pentavalent vaccine [10]. These vaccines are safe and effective, protecting thousands of individuals from contracting RV each year [11]. These vaccines may rarely result in an intestinal obstruction that occurs when a portion of the intestine folds back on itself (intussusception) [12]. In previous studies, de novo design was used for surveillance of vaccinations, which is crucial for detecting RV strains that are responsible for the spread of infections and tracking the evolution of the virus. Reassortant strains, both single and multiple, have been associated with these infections. Amino acid variations in the VP7 and VP4 antigens may contribute to vaccine escape [13, 14].
De novo protein design is the process of creating new proteins with sequences that do not occur in nature, based on the fundamental concepts of intramolecular and intermolecular interactions [15]. The computational methods were used to determine the accuracy achievable when designing a wide range of structures from scratch. The majority of protein design can alter the function of naturally occurring proteins [16]. These protein treatments are under development and testing. These treatments are derived from experiments aimed at desirable characteristics. Structure-based modeling allows for controlled alteration of the physical, chemical, and biological properties of proteins. This technology improves traditional rational engineering techniques by offering precise, hypothesis-driven pathways to identify protein issues and create innovative mechanisms of action [17-19].
Therefore, this study employed screening of de novo-designed proteins to target all strains of human RV proteins, which facilitate cell adhesion and virus penetration into host cells. These designed proteins can be considered novel antiviral targets of the human RV strains. The results of this research can help identify a new therapeutic strategy that prevents the proteins of the virus from binding with the host cell surface proteins and penetrating the host cells. However, further research is required to validate the findings of this study.
2. METHODOLOGY
2.1. Data Retrieving
An extensive literature review was conducted to retrieve data on all strains of RV. The protein sequences of the identified strains, provided in FASTA format, were subsequently searched in UniProtKB (https://www.uniprot. org/uniprotkb). UniProt is a comprehensive resource for protein function that provides accurate and detailed annotations of essential protein sequence data [20]. Subsequently, proteins present across all strains were identified. For each common protein, sequences from all strains were compiled into separate files. These files containing sequences of each protein from all strains were utilized for further analysis.
2.2. Multiple Sequence Alignment
Multiple sequence alignment (MSA) was performed to identify conserved regions of proteins across all RV strains within various species. The MSA was executed employing T-Coffee (https://tcoffee.crg.eu/apps/tcoffee/do:psicoffee). T-Coffee displays the conserved score for each species and the overall conserved score for all proteins [21]. T-Coffee results categorize alignments as good, bad, or average based on their accuracy in identifying homologous regions, reflecting the quality of sequence conservation and alignment. The overall conservation scores for all proteins, with 100 being the highest, were noted. Consequently, the top three proteins with the highest conserved scores were identified. Among these top conserved proteins, only rotavirus strains infecting humans were shortlisted for further analysis.
2.3. Structure Prediction of Conserved Proteins
Structures for shortlisted proteins with sequence lengths of less than 1000 amino acids were retrieved through the Omegafold. Moreover, proteins with greater amino acid length were predicted using SWISS-MODEL. OmegaFold leverages a novel protein language model and a geometry-aware transformer, achieving competitive accuracy with AlphaFold2 and surpassing RoseTTAFold for single-sequence protein structure prediction [22]. SWISS-MODEL develops a fully automated protein structure homology modeling server. It distributes a range of dependable, cutting-edge bioinformatics services and tools for the organization, analysis, and interpretation of biological data [23]. Lastly, secondary structures were predicted using SOPMA (Self-Optimized Prediction Method with Alignment), a secondary structure prediction tool that employs a query primary sequence of a protein to predict its secondary structure. Moreover, the tool analyzes and compares amino acid sequences with homologous sequences [24]. Three states, a window width of 17, and a similarity threshold of 8 were employed as default settings for secondary structure prediction.
2.4. Domain Analysis of Shortlisted Proteins
Domain analysis was carried out using the InterPro database (https://www.ebi.ac.uk/interpro/) to identify the domains of shortlisted proteins. The InterPro database identifies domains and their regions within protein structures. Additionally, the database curates a comprehensive protein classification system that integrates protein families, functional domains, and conserved sites for in-depth sequence analysis [25].
2.5. Identification and Screening of Interacting Proteins
The shortlisted proteins, acting through integrins or host proteins, mediate the incorporation of viruses into host cells, facilitating viral attachment and replication. Hence, an extensive literature review was conducted to identify integrins and other host proteins that interact with the shortlisted proteins.
The structures of identified interacting proteins were retrieved from the Protein Data Bank (https://www.rcsb. org/) and Alpha-Fold (https://alphafold.ebi.ac.uk/). The PDB structures were cleaned. Subsequently, the unmodelled regions of these structures were modeled through MODELLER (Comparative Protein Structure Modeling Software) [26]. Following the cleaning and modeling, these interacting proteins were screened against their respective shortlisted proteins through the ClusPro server (https://cluspro.org), a protein-protein docking tool that utilizes two pdb format files, offering advanced features like unstructured region removal, force modulation, and heparin-binding site identification [27, 28]. However, due to a server outage at ClusPro, the remaining screening was conducted through HDOCK (http://hdock.phys.hust.edu.cn/), a web server for template-based modeling and free docking that supports protein-protein, protein-DNA, and protein-RNA docking [29]. These screened protein-protein complexes were subsequently employed for further analysis.
2.6. Protein-protein Interaction Analysis
The PDBsum server (http://www.ebi.ac.uk/pdbsum) was used to analyze the molecular interactions in the three-dimensional models of the screened complexes generated by ClusPro and HDOCK. Using this tool, researchers can analyze bonded and non-bonded interactions between residues involved in protein-protein interactions [30]. Complex structures were visualized in PyMol (The PyMOL Molecular Graphics System, Version 4.5 (Compatibility Profile), running on Mesa 23.0.4-0ubuntu1~22.04.1) to provide cartoon representations, highlight interaction residues, and show polar interactions between docked protein complexes [31]. Moreover, interaction analysis was carried out to identify interactions within the domains of the shortlisted proteins.
2.7. De novo Protein Designing and Screening
To guide the de novo design process, the amino acid composition of the interaction interface for each shortlisted protein was determined from the PDBSum analysis. This residue frequency distribution was subsequently used as a compositional bias constraint in the ProteinGenerator tool (https://huggingface.co/spaces/merle/PROTEIN_GENERATOR). For each design cycle, a target protein length was predefined. The tool was then used to generate novel sequences that adhered to this specified amino acid bias, while all other optional parameters were kept at their default settings. The objective of this strategy was to generate new proteins whose amino acid makeup would mimic the native interaction sites. Subsequently, the designed protein sequences and their predicted 3D structures were filtered, and only high-confidence models, defined by a predicted Local Distance Difference Test (pLDDT) score greater than 0.75, were selected for further analysis. The thermal stability of the de novo designed protein sequences was predicted using the DeepStabP web server (https://csb-deepstabp.bio.rptu.de/). This server employs a deep learning model to estimate the stability score for each protein. To assess the potential toxicity of the designed proteins, the sequences were submitted to the CSM-Toxin web server (https://biosig.lab.uq.edu.au/ csm_toxin/). This tool uses machine learning to classify sequences as toxic or non-toxic.
The ClusPro server was utilized for the protein-protein docking between shortlisted protein structures and their respective de novo-designed proteins. Amongst all, the screened complexes with the highest binding affinities were identified. Subsequently, the interactions of these screened complexes within the domain regions of the shortlisted proteins were determined. Additionally, conservation of these interacting regions across all RV strains was observed.
3. RESULTS
3.1. Protein Sequence Retrieval of All Strains of Rotavirus
A total of nine RV strains were identified from the extensive literature review. The protein sequence data for each of the nine RV strains (A, B, C, D, F, G, H, I, and J), along with their FASTA sequences, were retrieved from UniProtKB. Additionally, strain sequences from different regions, such as the UK and China, as well as RV X, a substrain of the H strain, were retrieved. Common proteins, including RNA-directed RNA polymerase, outer capsid protein VP4, outer capsid glycoprotein VP7, non-structural protein 1, non-structural protein 2, non-structural protein 3, non-structural protein 5, non-structural glycoprotein 4, intermediate capsid protein VP6, inner capsid protein VP2, and VP3, were identified from each strain. The protein sequences of these 11 proteins were separated and compiled based on their function into separate files. Each protein file contained multiple protein FASTA sequences representing different strains of rotavirus from various species.
3.2. Multiple Sequence Alignment and Conserved Regions Analysis
T-Coffee was used to perform MSA analysis of the 11 aforementioned protein sequences to identify conserved regions within RV proteins. The MSA analysis results of 11 proteins are illustrated in the Supplementary Figs. (S1-S11). Amongst all, RNA-directed RNA polymerase, outer capsid VP4, and outer capsid glycoprotein VP7 proteins demonstrated the highest conservation of 85%, 77%, and 78%, respectively. Consequently, these three most conserved proteins were shortlisted for further analysis. The conserved score ratios (%) of all RV strain proteins are listed in Table 1.
3.3. Protein Structure Prediction
Following the MSA analysis, FASTA-format sequences of the shortlisted proteins from human RV strains only were retrieved, resulting in five sequences for each protein. These shortlisted interacting proteins of human rotavirus strains, along with their respective UniProt IDs, are listed in Table 2. Subsequently, structures of the VP7 and VP4 proteins were predicted using the Omegafold database, while RNA-directed RNA polymerase structures were predicted with SWISS-MODEL. The overview structures for all strains of the shortlisted proteins are illustrated in Fig. (1). The evaluation scores of these predicted structures are listed in Supplementary Sheet 1, Table S1.
| Protein | Conserved Region |
|---|---|
| Non-structural protein 3 | 69% |
| Non-structural protein 5 | 52% |
| Non-structural protein 1 | 51% |
| RNA-directed RNA polymerase | 84% |
| Intermediate capsid protein VP6 | 73% |
| Outer capsid glycoprotein VP7 | 78% |
| Non-structural protein 2 | 70% |
| Protein VP3 | 72% |
| Non-structural glycoprotein 4 | 40% |
| Outer capsid protein VP4 | 77% |
| Inner capsid protein VP2 | 65% |
Lastly, RNA-directed RNA polymerase proteins VP4 and VP7, and their secondary structures, were predicted using SOPMA. The analysis showed varying secondary structure elements in proteins, with the outer capsid proteins VP4 and VP7 having alpha-helix contents ranging from 30.78% to 36.52% and 27.31% to 35.58%, respectively. Notably, the RNA-directed RNA polymerase showed a higher alpha-helix content, ranging from 51.47% to 52.57%. Moreover, VP4 and VP7 had varying extended strand content, ranging from 17.81% to 26.91%, and RNA-directed RNA polymerase from 12.50% to 13.30%. The random coil ratios were high in all three proteins, ranging from 35% to 48%. Notably, no beta-turn structures were observed in any of the proteins analyzed. The secondary structures and their respective ratios in all strains of these three proteins are listed in Table 3. The secondary structures of all human strains of VP4, VP7, and RNA-directed RNA polymerase are illustrated in Fig. (2).
| Protein Names | UniProt ID | Strains |
|---|---|---|
| Outer capsid protein VP4 | J9QP39 | Human Rotavirus A |
| D6NGF7 | Human Rotavirus B (China) | |
| Q82040 | Human Rotavirus C (UK) | |
| Q32V57 | Human Rotavirus C | |
| Q45UF8 | Human Rotavirus X/ H (China) | |
| Outer capsid protein VP7 | Q0GC14 | Human Rotavirus A |
| D6NGF9 | Human Rotavirus B (China) | |
| Q89865 | Human Rotavirus C (UK) | |
| Q32UE9 | Human Rotavirus C | |
| Q45UF2 | Human Rotavirus X/H (China) | |
| RNA-directed RNA polymerase | A0A059P831 | Human Rotavirus A |
| D6NGF4 | Human Rotavirus B (China) | |
| Q91E95 | Human Rotavirus C (UK) | |
| E5KJF9 | Human Rotavirus C | |
| Q45UG0 | Human Rotavirus X/H (China) |
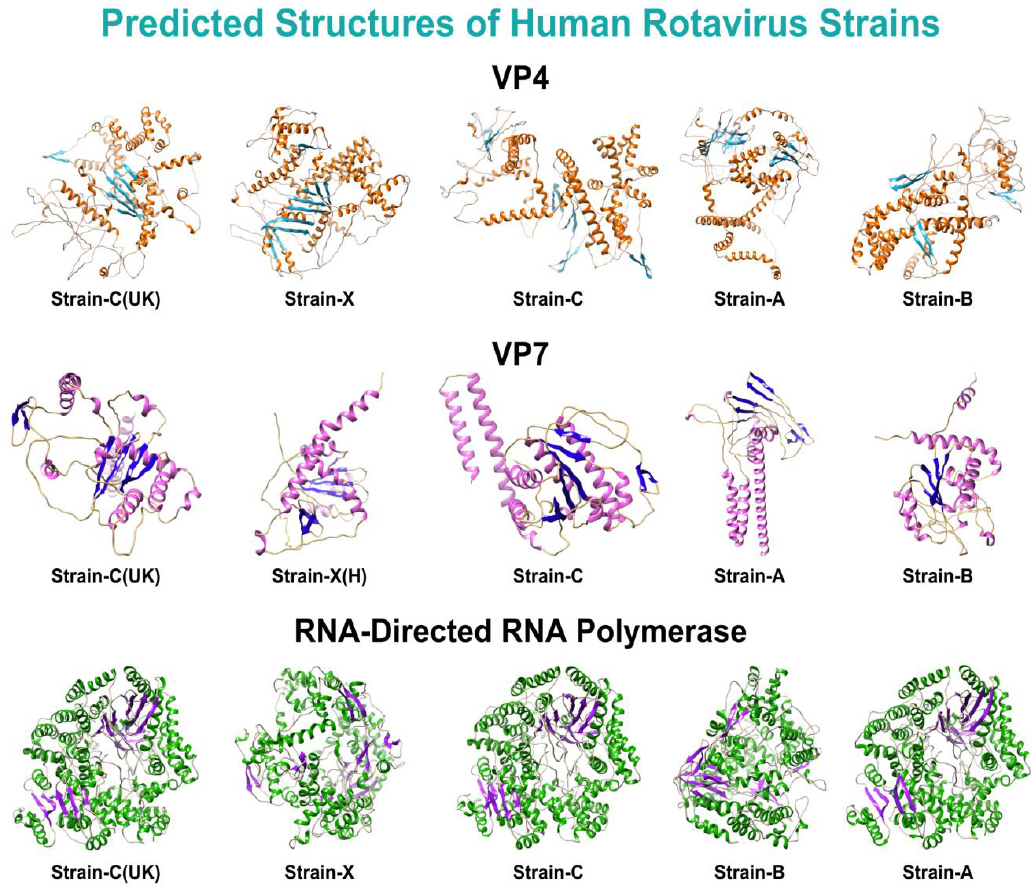
Predicted structures of all strains of the shortlisted proteins.
| Proteins | Strains | Alpha Helix | Extended Strand | Beta Turn | Random Coil |
|---|---|---|---|---|---|
| Outer capsid protein VP4 | Human Rotavirus A | 36.52% | 17.81% | 0.00% | 45.68% |
| Human Rotavirus B (China) | 35.07% | 18.67% | 0.00% | 46.27% | |
| Human Rotavirus C (UK) | 30.78% | 20.56% | 0.00% | 48.66% | |
| Human Rotavirus C | 33.87% | 18.55% | 0.00% | 47.58% | |
| Human Rotavirus X/H (China) | 32.20% | 19.08% | 0.00% | 48.72% | |
| Outer capsid protein VP7 | Human Rotavirus A | 35.58% | 21.78% | 0.00% | 42.64% |
| Human Rotavirus B (China) | 27.31% | 26.91% | 0.00% | 45.78% | |
| Human Rotavirus C (UK) | 28.92% | 23.19% | 0.00% | 47.89% | |
| Human Rotavirus C | 31.33% | 23.80% | 0.00% | 44.88% | |
| Human Rotavirus X/H (China) | 27.52% | 26.74% | 0.00% | 45.74% | |
| RNA-directed RNA polymerase | Human Rotavirus A | 52.57% | 12.50% | 0.00% | 34.93% |
| Human Rotavirus B (China) | 51.55% | 12.59% | 0.00% | 35.86% | |
| Human Rotavirus C (UK) | 51.74% | 13.21% | 0.00% | 35.05% | |
| Human Rotavirus C | 51.47% | 13.30% | 0.00% | 35.23% | |
| Human Rotavirus X/H (China) | 52.10% | 13.02% | 0.00% | 34.88% |
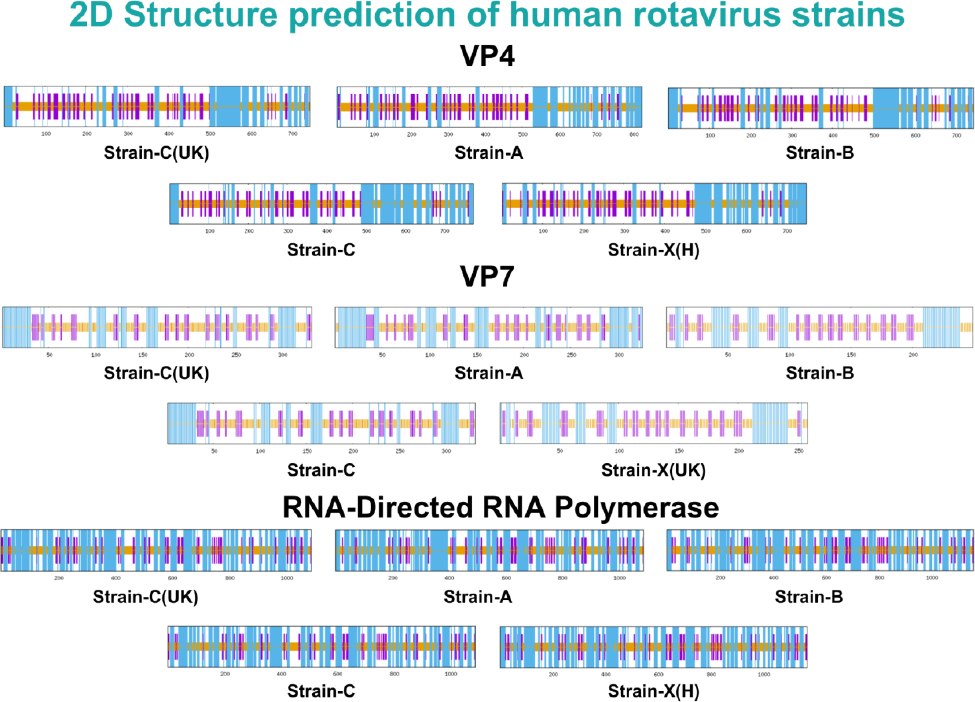
Secondary structures of all strains of the shortlisted proteins.
3.4. Domain Analysis of Shortlisted Proteins
A domain analysis of the shortlisted proteins was conducted using the InterPro database to identify key functional regions. The results, summarized in Table 4, reveal significant architectural differences among human rotavirus strains, as visualized in the superimposed structures shown in Fig. (3).
| Outer capsid VP4 | |
|---|---|
| Protein Name | Domain Name |
| Human rotavirus strain A and both C strains | 1) Rotavirus VP4 helical domain 2) Haemagglutinin outer capsid protein VP4 concanavalin-like domain 3) Rotavirus VP4, membrane interaction domain |
| Human rotavirus B and X/H | 1) Rotavirus VP4, membrane interaction domain |
| Outer capsid VP7 | |
| Protein Name | Domain Name |
| Human rotavirus strain A and both C strains | 1) Glycoprotein VP7, domain 1 |
| Human rotavirus B and X/H | No Domain |
| RNA-directed RNA polymerase | |
| Protein Name | Domain Name |
| Human rotavirus strain A and both C strains | 1) RNA-directed RNA polymerase, reovirus 2) Rotavirus VP1 RNA-directed RNA polymerase, C-terminal |
| Human rotavirus B and X/H | 1) RNA-directed RNA polymerase, reovirus |
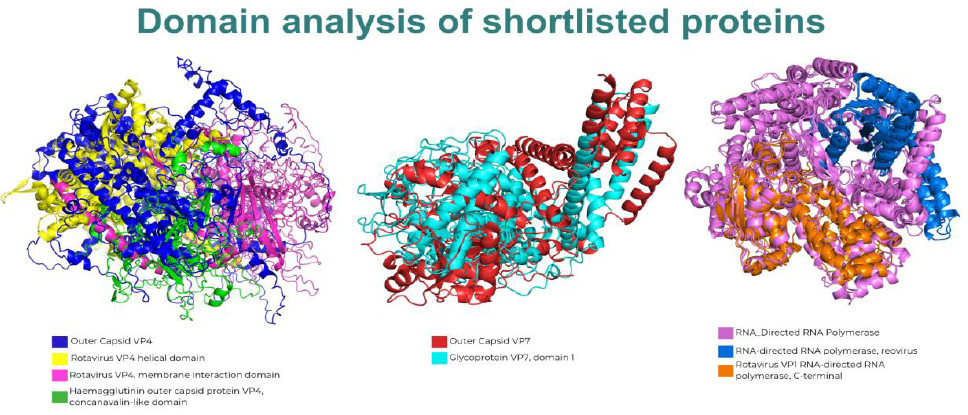
Comparative Domain Architecture of Shortlisted Rotavirus Proteins. Superimposed 3D structures of all analyzed human rotavirus strains for the outer capsid VP4 (left), outer capsid VP7 (center), and RNA-directed RNA polymerase (right), highlighting the presence or absence of key functional domains across different strains. For VP4, the composite structure shows the helical domain (yellow), membrane interaction domain (green), and concanavalin-like domain (pink) in strains A/C, whereas strains B/X/H contain only the membrane interaction domain. For VP7, a distinct glycoprotein VP7 domain 1 (cyan) is visible for strains A/C but is absent in strains B/X/H. Finally, for RNA-directed RNA polymerase, strains A/C feature both the reovirus domain (blue) and a C-terminal domain (orange), whereas the C-terminal domain is missing in strains B/X/H.
For the outer capsid VP4 protein, strains A and C were found to contain three distinct domains: the Rotavirus VP4 helical domain, the Haemagglutinin outer capsid protein VP4 (concanavalin-like) domain, and the rotavirus VP4 membrane interaction domain. In contrast, strains B and X/H possess only the rotavirus VP4 membrane-interaction domain. This difference is visually represented in Fig. (3) (left panel), where the composite structure highlights the multiple domains contributed by strains A and C, whereas strains B and X/H map only to the singular membrane interaction region.
Analysis of the outer capsid VP7 protein revealed a more pronounced divergence. As shown in the central panel of Fig. (3), strains A and C harbor a distinct VP7 domain 1, which is absent in strains B and X/H. This observation clearly illustrates a significant structural difference between these sets of strains.
Finally, the RNA-directed RNA polymerase also showed strain-specific domain compositions. Strains A and C exhibit two domains: the RNA-directed RNA polymerase (reovirus) domain and a C-terminal domain. However, strains B and X/H lack the C-terminal domain. Figure 3 (right panel) intuitively reflects this finding, showing the complete two-domain structure for strains A and C, whereas strains B and X/H contain only the reovirus domain.
3.5. Identification and Screening of Interacting Proteins
An extensive literature review was conducted using the Google Scholar and PubMed databases to identify proteins that interact with the shortlisted proteins (VP4, VP7, and RNA-directed RNA polymerase) implicated in infection. Outer capsid VP4 was reported to interact with integrins α2β1 and α4β1, whereas VP7 interacted with integrins αxβ2, α4β1, and αVβ3. Moreover, human infection was reported to be facilitated by the interaction between RNA-directed RNA polymerase and its inner capsid protein, VP2. The literature review was performed using both subject terms and free words, such as “VP4 interacting proteins,” “VP7 interacting proteins,” “RNA-directed RNA polymerase interacting proteins,” “Rotavirus host protein interaction,” and “VP4/VP7/RNA polymerase binding partners in human cells.” These identified interacting proteins were then utilized for further analysis. The structures of these identified interacting proteins were retrieved from pdb. However, the structural prediction of the inner capsid protein VP2 (UniProt ID Q86195) was performed using the SWISS-MODEL. The identified human-interacting proteins, along with their respective PDB IDs, are listed in Table 5.
| Rotavirus Proteins | Interaction Protein | Protein Names | PDB IDs |
|---|---|---|---|
| Outer capsid protein VP4 | α2β1 | Integrin alpha-2/beta-1 | ITGA2 |
| α4β1 | Integrins alpha-4/beta-1 | 3V4P | |
| Outer capsid protein VP7 | αxβ2 | Integrin alpha-X/beta-2 | 3K6S |
| α4β1 | Integrins alpha-4/beta-1 | 3V4P | |
| αVβ3 | Integrin alpha-V/beta-3 | ITGB3 | |
| RNA-directed RNA polymerase | VP2 | Inner capsid protein VP2 | - |
The different strains of shortlisted proteins were screened against their respective interacting proteins. Protein-protein docking analysis using ClusPro was performed, yielding 10 complexes for VP4, 10 for VP7, and 5 for RNA-directed RNA polymerase. Among VP4 complexes, the highest binding affinity of -2034.6 was observed for strain B with ITGA2 (α2β1), whereas the lowest affinity of -1235 was observed for the interaction of strain B with 3VAP (α4β1). For VP7 complexes, the highest affinity of -1808.6 was observed between strain C VP7 and 3V4P (α4β1), while the lowest affinity of -1296 was observed between strain B and 3K6S (αxβ2). In RNA-directed RNA polymerase complexes, the highest affinity of -1055 was observed between strain A and VP2, whereas the lowest affinity of -1486.9 was seen with strain B and VP2. The results of these 25 complexes are mentioned in Table 6. However, due to the server outage on ClusPro, VP7 strains were screened for ITGB3 (αVβ3) using HDOCK. The highest docking score of -256.75 was observed for strain B with ITGB3, while the lowest score was -307.61 observed for strain C with ITGB3. The screening HDOCK results of the 5 complexes are listed in Table 7. These 30 screened complexes were then utilized for further analysis.
| Complex | Affinities |
|---|---|
| VP4_Human rotavirus A_ITGA2 | -1759.9 |
| VP4_Human rotavirus A_3VAP | -1582.9 |
| VP4_Human rotavirus B_ITGA2 | -2034.6 |
| VP4_Human rotavirus B_3VAP | -1253 |
| VP4_Human rotavirus C (UK)_ITGA2 | -1686.6 |
| VP4_Human rotavirus C (UK)_3VAP | -1340.9 |
| VP4_Human rotavirus C_ITGA2 | -1980.3 |
| VP4_Human rotavirus C_3VAP | -1483.4 |
| VP4_Human rotavirus X/H_ITGA2 | -1644.1 |
| VP4_Human rotavirus X/H_3VAP | -1562.5 |
| VP7 Human rotavirus A_3V4P | -1408.9 |
| VP7 Human rotavirus A_3K6S | -1524 |
| VP7 Human rotavirus B_3V4P | -1341.1 |
| VP7 Human rotavirus B_3K6S | -1296 |
| VP7 Human rotavirus C (UK)_3V4P | -1532.3 |
| VP7 Human rotavirus C (UK)_3K6S | -1553.8 |
| VP7 Human rotavirus C_3V4P | -1808.6 |
| VP7 Human rotavirus C_3K6S | -1551.6 |
| VP7 Human rotavirus X/H_3V4P | -1372.2 |
| VP7 Human rotavirus X/H_3K6S | -1274 |
| RNA-directed RNA polymerase_Human rotavirus A_VP2 | -1055 |
| RNA-directed RNA polymerase_Human rotavirus B_VP2 | -1486.9 |
| RNA-directed RNA polymerase_Human rotavirus C (UK)_VP2 | -1196.9 |
| RNA-directed RNA polymerase_Human rotavirus C_VP2 | -1212.1 |
| RNA-directed RNA polymerase_Human rotavirus X/H_VP2 | -1368.2 |
| Complex | Docking Score | Confidence Score |
|---|---|---|
| VP7 Human rotavirus A_ITGB3 | -278.73 | 0.9292 |
| VP7 Human rotavirus B_ITGB3 | -256.75 | 0.8943 |
| VP7 Human rotavirus C (UK)_ITGB3 | -268.12 | 0.9139 |
| VP7 Human rotavirus C_ITGB3 | -307.61 | 0.959 |
| VP7 Human rotavirus C_ITGB3 | -281.84 | 0.9332 |
3.6. Protein-protein Interactions Analysis
Using PDBsum1, residue-wise analysis of protein-protein interactions in 30 screened complexes was carried out, revealing an extensive range of interactions, along with their corresponding residues and positions across all complexes. The PDBSum analysis results for these shortlisted protein complexes are listed in Supplementary Sheet 1 (Table S2-S4, respectively). Among the complexes analyzed, some showed stronger interactions than others. Outer capsid VP4 human RV strains A and B exhibited interactions with both α4β1 and α2β1 integrins within multiple domains. Strain A exhibited 15 interactions with α4β1 within the rotavirus VP4 helical domain (485-775) and 8 interactions with α2β1 within the rotavirus VP4 membrane interaction domain (249-473). Conversely, strain B exhibited 15 interactions with α2β1 within the rotavirus VP4 membrane interaction domain superfamily. Similarly, C strains exhibited 28 interactions with α2β1 and α4β1 within the Haemagglutinin outer capsid protein VP4, the concanavalin-like domain (71-234), the rotavirus VP4 membrane interaction domain (260-486), and the rotavirus VP4 helical domain (497-744). Lastly, strain X/H exhibited 4 interactions with α2β1 and 2 interactions with α4β1 within the VP4 membrane-interaction domain (residues 272–394) of rotavirus.
Outer capsid VP7 human RV strain A did not show interactions with α4β1 but exhibited interactions with αxβ2 and αVβ3 integrins. Notably, αxβ2 showed 9 interactions, including one with αVβ3, within domain 1 (residues 77–316) of the VP7 glycoprotein. However, strain B and strain X/H exhibited no interactions with α4β1. Moreover, no domain was reported for these strains. Strain C demonstrated interactions with α4β1, αxβ2, and αVβ3 within the glycoprotein VP7 domain 1 (77-316). The α4β1 exhibited 11 interactions, while αxβ2 exhibited 9 interactions, and αVβ3 exhibited 4 interactions.
The RNA-directed RNA polymerase of strain A exhibited nine interactions, and that of strain B exhibited four interactions with VP2 within residues 501–687 and 546–740 of the reovirus RNA-directed RNA polymerase. Notably, strain C exhibited 22 interactions within the C-terminal domain (residues 778–1090) and the RNA-directed RNA polymerase domain (residues 506–678) of the reovirus RNA-directed RNA polymerase, whereas strain X/H exhibited no interactions. The interactions between proteins, their interacting partners, and the corresponding residues and domains are detailed in Table 8.
| Outer capsid VP4 | |||
|---|---|---|---|
| Protein | Interacting Proteins | Interacting Residues | Domain |
| Human, Rotavirus A | α4β1 | TYR (614), SER (616), LYS (617), GLN (624), ARG (632), TYR (644), ASN (649), ARG (656), TYR (664), ASP (667), ASP (669), HIS (672), GLU (673), SER (676), LYS (693) | 1) Rotavirus VP4 helical domain (485-775) 2) Rotavirus VP4, membrane interaction domain (249-473) |
| α2β1 | LYS (295), TYR (299), ARG (361), ARG (367), SER (368), ALA (371), SER (463), GLY (464) | ||
| Human, Rotavirus B (China) | α4β1 | No interactions | 1) Rotavirus VP4, membrane interaction domain superfamily (216-513) |
| α2β1 | LYS (240), TYR (267), MET (268), HIS (277), ASP (311), TYR (317), ARG (363), GLY (399), GLU (400), ASP (413), THR (416), TYR (425), CYS (438), CYS (439), GLU (580) | ||
| Human Rotavirus C (UK) | α4β1 | ARG (80) | 1) Haemagglutinin outer capsid protein VP4, concanavalin-like domain (71-234) |
| Human Rotavirus C | α2β1 | THR (281), GLN (283), CYS (284), GLY (297THR (307), ARG (335), GLN (387), VAL (438), THR (450), ARG (457), SER (461), THR (463), PHE (465), ASP (279), TYR (298), TYR (300), SER (340), TYR (363), MET(373), GLU(393), SER (395), GLN(414), LYS(180), GLU(197), ASN(200), ARG(205), ASP(279) | 1) Haemagglutinin outer capsid protein VP4, concanavalin-like domain (71-234) 2) Rotavirus VP4, membrane interaction domain (260-486) 3) Rotavirus VP4 helical domain (497-744) |
| α4β1 | SER (262), ILE (265), LYS (267), ARG (502)ASP (231), GLN (234)ARG (502), ARG (513), ASN (517), LYS (562), LYS (571), GLN (575), ASN (576), GLU (579) | ||
| Human, Rotavirus X/H (China) | α4β1 | LYS (274), ARG (374) | 1) Rotavirus VP4, membrane interaction domain (272-394) |
| α2β1 | SER (362), HIS (363), ASN (364), ASP (365) | ||
| Outer capsid VP7 | |||
| Protein | Interacting Proteins | Interacting Residues | Domain |
| Human Rotavirus A | α4β1 | No interactions | Glycoprotein VP7, domain 1 (79-312) |
| αxβ2 | THR (112), TYR (116), THR (275), MET (277), GLN (279), TRP (291), TYR (296), ASN (303), GLN (307) | ||
| αVβ3 | GLN (279) | ||
| Human Rotavirus B (China) | α4β1 | No interactions | No Domain Observed |
| Human Rotavirus C (UK) | α4β1 | TRP (162), ASP (171), ASN (173), GLU (175), SER (271), ARG (329) | Glycoprotein VP7, domain (77-316) |
| αxβ2 | MET (172, LEU (174), THR (178), GLU (180), GLN (181), THR (226), ASN (275), LYS (290), LYS (294), TYR (301) | ||
| αVβ3 | GLN (181), ASN (275) | ||
| Human Rotavirus C | α4β1 | ASN (101), ASN (101), THR (105), LYS (113), LYS (117) | Glycoprotein VP7, domain 1 (77-316) |
| αxβ2 | No interactions | ||
| αVβ3 | HIS (239), ARG (319) | ||
| Human Rotavirus X/H (China) | α4β1 | No interactions | No Domain Observed |
| RNA-directed RNA polymerase | |||
| Protein | Interacting Proteins | Interacting Residues | Domain |
| Human Rotavirus A | Inner capsid protein VP2 | ARG (614, ASN (617), LYS (618), TYR (619), SER (620), THR (623), LYS624), THR (641), GLN (646) | 1) RNA-directed RNA polymerase, reovirus (501-687) |
| Human, Rotavirus B (China) | SER (630), GLY (632), LYS (638), ARG (641) | 1) RNA-directed RNA polymerase, reovirus (546-740) | |
| Human, Rotavirus C (UK) | ASN (780), TYR (880), PHE (885), LYS (887), LYS (984), SER (1057), ASN (1061), LYS (1062), ASP (1064), LYS (1067), ARG (1070), TRP (1073), ASN (1074) | 1) RNA-directed RNA polymerase, C-terminal (778-1090) | |
| Human, Rotavirus C | ARG (78), ASN (780), GLY (884), ASN (954), HIS (1059, ASN (1061), THR (1063), LYS (1067), ARG (1070), TRP (1073), ASN (1074) | 1) RNA-directed RNA polymerase, reovirus (506-678) | |
| Human rotavirus X/H (China) | No interactions | 1) RNA-directed RNA polymerase, reovirus (553-735) | |
3.7. De Novo Protein Designing and Docking
Fraction ratios for the identified interactions were calculated from residue-wise analyses of interacting proteins. These fraction ratios of interacting proteins for their respective shortlisted proteins are mentioned in Supplementary Sheet 1 (Tables S5-S7). These fraction ratios were utilized to design a de novo protein for each complex. Amongst all, the top 5 de novo-designed proteins (D1, D2, D3, D4, and D5) were selected based on the model confidence score. ClusPro was used to screen these five de novo-designed proteins against all strains of the respective human RV proteins (VP4, VP7, and RNA-directed RNA polymerase). This resulted in 25 complexes per protein, each representing the interaction between each strain and the five de novo proteins. The screening results of these complexes are listed in Supplementary Sheet 1 (Tables S8-S10). The computational analysis of thermal stability revealed high predicted stability scores across all three protein groups. The mean stability scores were 73.38 for the RNA polymerase group, 73.15 for the VP4 group, and 74.22 for the VP7 group. The distribution of these scores, as shown in the boxplot, indicates that all three groups have similar and favorable stability profiles. The toxicity predictions from CSM-Toxin showed that the vast majority of the designed proteins were non-toxic. Out of 89 sequences analyzed, 86 (96.6%) were predicted to be non-toxic, while only 3 were classified as toxic. Specifically, the VP4 group was predicted to be entirely non-toxic, whereas one sequence from the RNA polymerase group and two from the VP7 group were flagged as potentially toxic (Supplementary Figs. S12-S13).
Screening outer capsid VP4 human RV strains against their de novo-designed proteins yielded binding affinities ranging from -854.5 to -1769.2. Amongst all de novo-designed proteins, the D1-designed protein for strains A and B exhibited the highest binding affinities of -1769.2 and -1358.2, respectively. Similarly, the D4-designed protein for both C strains exhibited the highest binding affinities of -1661.8 and -1334.1, respectively. Lastly, the D2-designed protein for strain X/H exhibited a high binding affinity of -1339.9. Notably, the interaction sites for these strains were conserved across all species. Moreover, all interactions occurred within the rotavirus VP4 membrane interaction domain.
Subsequently, screening outer capsid VP7 human RV strains against their de novo-designed proteins yielded binding affinities ranging from -687.3 to -1608.7. Notably, amongst all de novo-designed proteins, the D5-designed protein for A, B, and both C strains exhibited the highest binding affinities of -1355.3, -1120.5, -1127.3, and -1608.7, respectively. Moreover, the D1-designed protein for the X/H strain exhibited the highest binding affinity of -1264.1. The interaction sites for only X/H, B, and C strains were conserved across all species. In contrast, interactions involving only A and both C strains occurred within the glycoprotein VP7 domain 1.
Lastly, screening RNA-directed RNA polymerase strains against their de novo-designed proteins yielded binding affinities ranging from -602.6 to -1378.9. Amongst all de novo-designed proteins, the D5-designed protein for strains A and C exhibited the highest binding affinities of -846.6 and -812.2, respectively. Similarly, the D2-designed protein for strains B and C (UK) exhibited the highest binding affinities of -1378.9 and -815.6, respectively. Lastly, the D4-designed protein for strain X/H exhibited the highest binding affinity of -1049.4. Notably, the interaction sites for these strains were conserved across all species. Moreover, interactions involving only A and both C strains occurred within the C-terminal domain of RNA-directed RNA polymerase. In contrast, the interactions of strain B occurred within the RNA-directed RNA polymerase domain of the reovirus. However, strain X/H did not interact within any domain. The results for the highest-affinity complexes, along with their corresponding interaction domains, are listed in Table 9.
| Outer capsid VP4 | |||
|---|---|---|---|
| Strain | De Novo Protein | Affinities | Domain |
| Strain A | D1 | -1769.2 | Rotavirus VP4, membrane interaction domain |
| Strain B | D1 | -1358.2 | |
| Strain C | D4 | -1661.8 | |
| Strain C (UK) | D4 | -1334.1 | |
| Strain X/H | D2 | -1339.9 | |
| Outer capsid VP7 | |||
| Strain | De Novo Protein | Affinities | Domain |
| Strain A | D5 | -1355.3 | Glycoprotein VP7, domain 1 |
| Strain B | D5 | -1120.5 | Outside domain |
| Strain C | D5 | -1127.3 | Glycoprotein VP7, domain 1 |
| Strain C (UK) | D5 | -1608.7 | |
| Strain X/H | D1 | -1264.1 | Outside domain |
| RNA-Directed RNA Polymerase | |||
| Strain | De Novo Protein | Affinities | Domain |
| Strain A | D5 | -846.6 | RNA-directed RNA polymerase, C-terminal |
| Strain B | D2 | -1378.9 | RNA-directed RNA polymerase, reovirus |
| Strain C | D5 | -812.2 | RNA-directed RNA polymerase, C-terminal |
| Strain C (UK) | D2 | -815.6 | |
| Strain X/H | D4 | -1049.4 | Outside domain |
Furthermore, the interactions occurring within the conserved regions are illustrated in Fig. (4).
4. DISCUSSION
RV, a member of the family Sedoreoviridae, causes acute to severe gastroenteritis and diarrhea in infants and children worldwide [1]. Notably, this disease is reported as a fatal disease due to high death rates in low-income countries [3]. RV is a double-stranded RNA with eight strains: A, B, C, D, E, F, G, and H, and each strain further has six structural (VP1, VP2, VP3, VP4, VP6, and VP7) and six non-structural (NSP1-NSP6) proteins [32]. These proteins help the RV penetrate and replicate its DNA in the host's body.
Oral vaccines are less effective at protecting against infection in low-income countries [33]. Strain diversity, the low immunogenicity of oral vaccines, and difficulties in targeting the population may explain the low efficacy of licensed vaccines in low-income countries. The efficiency of these vaccines can be increased by overcoming these limitations [34]. Developing traditional vaccines is a complex, costly, time-consuming, and labor-intensive process [35]. Hence, more efficient methods are required to replace these traditional strategies.
Notably, the de novo protein design strategy has been reported to be effective at generating highly regular structures with specified structural and functional properties [36]. This technique shows great potential for developing highly effective vaccines that can replicate a viral antigen in the absence of the natural protein. These designed proteins have shown the potential to target virus-neutralizing antibodies (nAbs) in vivo. Therefore, in this study, de novo protein designing is employed to target all human RV strains of the three most conserved proteins (VP7, VP4, and RNA-directed RNA polymerase). These proteins play a crucial role in the replication and transmission of viral DNA within the host. The RNA-directed RNA polymerase is reported to assist the replication of viral RNA [1, 37]. Similarly, the outer capsid proteins VP4 and VP7 bind receptors and help the virus enter the host cell [1]. Furthermore, some studies have employed de novo design for targeting non-structural protein 5 (NSP5) and non-structural protein 2 (NSP2) of RV [38, 39].
Notably, α2β1, α4β1, αxβ2, αVβ3, and VP2-interacting proteins have been reported to interact with human RV strains. The α2β1 and α4β1 integrins exhibited interactions with VP4, whereas VP7 interacted with αxβ2, α4β1, and αVβ3 [8, 40-42]. Similarly, the VP2 protein was found to be reported to interact with the RNA-directed RNA polymerase. Similarly, in this study, the outer capsid VP4 of human RV strains A and B interacted with α4β1 and α2β1 integrins across multiple domains, including the rotavirus VP4 helical domain and the rotavirus VP4 membrane interaction domain. Moreover, the outer capsid VP7, strain A, and both C strains interacted with αxβ2, αVβ3, and α4β1 integrins within the glycoprotein VP7 domain 1. The binding motifs of VP4 and VP7 are crucial for cellular interaction, fusion of viral and host cell membranes during viral entry, and membrane penetration during the viral life cycle, primarily mediating viral entry into host cells [43]. Furthermore, interactions of the RNA-directed RNA polymerase varied among different strains. Strains A, B, and C displayed interactions with VP2 within the RNA-directed RNA polymerase C-terminal and reovirus domains. The VP2 protein was reported to facilitate replication by interacting with the RNA-directed RNA polymerase, resulting in infection [44]. This highlights the need to inhibit binding interactions, thereby preventing the virus from causing infections.
Consequently, in this study, the five de novo-designed proteins (D1, D2, D3, D4, and D5) were screened against all strains of their respective human RV proteins (VP4, VP7, and RNA-directed RNA polymerase) to target the aforementioned interacting sites involved in mediating the virus into the host, leading to RV infection. Notably, the D1- and D4-designed proteins for VP4 interacted with A, B, and C strains within the rotavirus VP4 membrane interaction domain. Moreover, this interaction site was conserved across all strains. Similarly, the D5-designed protein for VP7 interacted with only A and C strains within the VP7 glycoprotein domain 1. Additionally, the D1-designed protein interacted with the X/H strains outside the domain. The interaction sites for only the X/H and C strains were conserved across all species. Lastly, the D5, D2, and D4-designed proteins for RNA-directed RNA polymerase interacted with all strains. The interactions of all strains, except the X/H strain, occurred within the RNA-directed RNA polymerase C-terminal domain and the RNA-directed RNA polymerase reovirus domain and showed conservation across all species. Notably, these findings highlight multiple interactions between the de novo-designed proteins and RV human proteins occurring within similar domain regions. The results of our computational analyses provide strong evidence that the de novo-designed proteins are both structurally stable and generally non-toxic. The high thermal stability scores predicted by DeepStabP suggest that the designed sequences are likely to fold into stable three-dimensional structures, a critical requirement for their intended function.
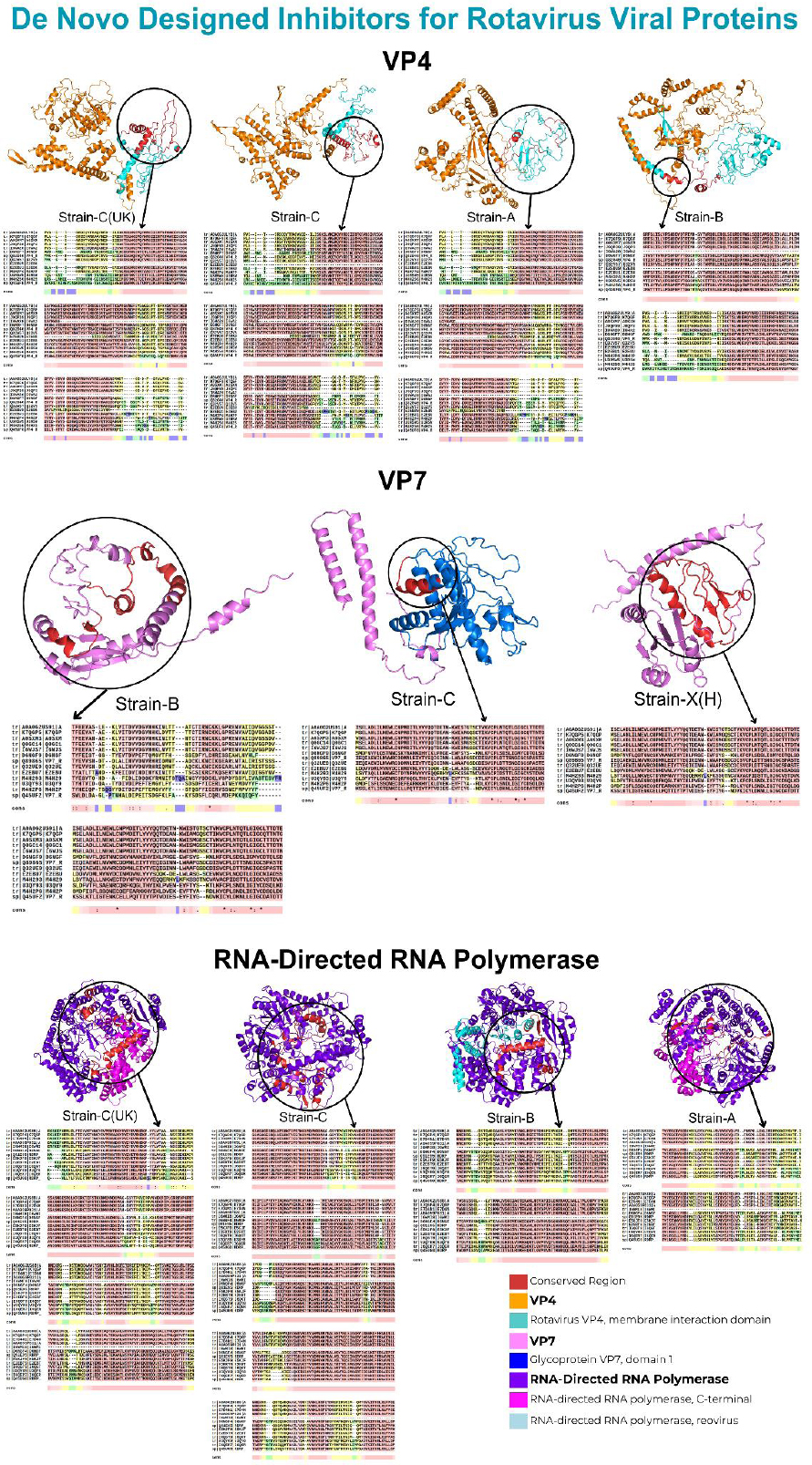
Interactions within conserved regions of human RV proteins.
Furthermore, the low predicted toxicity is a crucial finding for the potential downstream applications of these proteins. The fact that over 96% of the sequences were classified as non-toxic indicates that our design methodology is effective at generating safe and viable protein candidates. Identifying a small number of potentially toxic sequences enables their exclusion from further experimental validation, thereby streamlining the development process. Overall, these computational results support the success of our de novo protein design approach and provide a solid foundation for future experimental characterization.
A critical aspect of validating our in-silico findings is to compare them with established biological data. Our docking analysis identified several key interaction residues, including TYR614 and SER616, in the VP4 protein. When mapped onto the known architecture of the rotavirus spike protein, many of these predicted contact points fall within or are spatially proximal to recognized functional domains. For example, the VP4 helical domain, where these residues are located, undergoes significant conformational changes that are essential for membrane penetration during viral entry. The proximity of our predicted residues to these functionally critical regions lends biological plausibility to our model. Therefore, these designed proteins can be considered significant inhibitors, preventing the virus from penetrating the host cell. This de novo protein design can be considered a novel therapeutic strategy for combating RV infection in humans.
5. STUDY LIMITATIONS
While this study offers promising insights into de novo protein design targeting conserved domains of rotavirus proteins, several limitations should be acknowledged.
First, the research is entirely computational and lacks experimental validation. Although docking and structural prediction tools (e.g., ClusPro, HDOCK, SOPMA, OmegaFold, SWISS-MODEL) provide robust predictions, in vitro and in vivo assays are necessary to confirm the biological activity, specificity, and inhibitory effects of the designed proteins against rotavirus.
Second, the variability in binding affinities across different strains and the lack of domain-specific interactions in some designed complexes, particularly in strains like X/H, suggest that the designed proteins may have variable efficacy across the full spectrum of circulating human rotavirus strains.
Third, the study relies on existing databases (UniProtKB, PDB, AlphaFold) and on the accuracy of domain prediction tools, such as InterPro. These tools may have limitations in annotating novel or less-characterized proteins, potentially affecting the interpretation of interaction domains.
Fourth, host factors, such as immune response, protein degradation, and intracellular trafficking, were not considered in the current analysis, which could significantly affect the stability and efficacy of these designed therapeutic candidates.
Lastly, this study does not address delivery mechanisms, immunogenicity, or potential off-target effects of the designed proteins. These aspects are crucial for translating computational designs into clinically applicable antivirals or vaccine candidates.
Future studies should focus on experimental validation, including biophysical interaction studies, cell-based assays, and animal models, to validate the inhibitory potential and therapeutic efficacy of the designed proteins. Additionally, structural refinement, solubility enhancement, and immunogenicity testing will be necessary to transition from in silico design to clinical application.
CONCLUSION
Conclusively, this study investigates the de novo design strategy to target human RV strains of the VP4, VP7, and RNA-directed RNA polymerase proteins. These proteins play a crucial role in cell attachment and binding, as well as in the virus's penetration into the host cell. Notably, the de novo-designed proteins exhibited multiple interactions with the aforementioned proteins, primarily in the rotavirus VP4 membrane interaction domain, the glycoprotein VP7 domain 1, the RNA-directed RNA polymerase C-terminal domain, and the RNA-directed RNA polymerase reovirus domains. These findings underscore the potential of de novo proteins as effective inhibitors against the aforementioned proteins involved in RV infection. However, further clinical investigations are necessary to evaluate the effectiveness of these inhibitors in combating RV infections.
AUTHORS’ CONTRIBUTIONS
The authors confirm contribution to the paper as follows: N.B.A. and N.M.A.: Performed data collection, analysis, and literature review; A.A.S.: Supervised the project, contributed to the design and methodology, and reviewed the manuscript. All authors read and approved the final manuscript.
LIST OF ABBREVIATIONS
| RV | = Rotavirus |
| VP | = Viral Protein |
| NSP | = Non-Structural Protein |
| MSA | = Multiple Sequence Alignment |
| PDB | = Protein Data Bank |
| SOPMA | = Self-Optimized Prediction Method with Alignment |
| MODELLER | = Comparative Protein Structure Modeling Software |
| HDOCK | = Hybrid Docking Web Server |
| PDBsum | = Protein Data Bank Summary |
| DLP | = Double-Layered Particle |
| nAbs | = Neutralizing Antibodies |
| AA | = Amino Acid |
AVAILABILITY OF DATA AND MATERIALS
All data generated or analyzed during this study are included in this published article and its supplementary information files. Additional datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
ACKNOWLEDGEMENTS
The authors would like to acknowledge the support provided by the University of Anbar and Tikrit University. They would also like to thank the Bioinformatics Unit team for their technical assistance and insights throughout the analysis.

