All published articles of this journal are available on ScienceDirect.

Heat Shock Protein 90 (HSP90) as a Universal Target in Rhizopus Strains: Overcoming Antifungal Resistance through Bioinformatics
Abstract
Introduction
In an attempt to solve the problem of antifungal resistance in the Rhizopus species, which contributes to the severity of mucormycosis, the study was conducted to target the Heat Shock Protein 90 (HSP90). The main aim was to develop a new de novo protein inhibitor that was specific to the fungal HSP90 but not to the human counterpart, reducing off-target toxicity, and overcoming the limitations of the current therapies.
Methods
A bioinformatics approach was adopted. The strategy involved determining the conserved domains of the HSP90 protein of different strains of Rhizopus through multiple sequence alignment. The ten possible de novo protein inhibitors were then generated using a deep learning model on the basis of the consensus sequence of this conserved region. The stability and binding affinity of these inhibitors were measured through molecular dynamics simulations and protein-protein molecular docking to the fungal and human HSP90 structures.
Results
The analysis led to the identification of an inhibitor of lead de novo, Gen7, which exhibited improved binding and specificity. Molecular docking revealed that Gen7 had much higher affinity and interacted extensively with Rhizopus stolonifer HSP90 (12 hydrogen bonds, 4 salt bridges) than with human HSP90 (5 hydrogen bonds, 3 salt bridges). This fungal selectivity was later confirmed by subsequent molecular dynamics simulations. The Gen7-R. stolonifer complex was highly stable with an RMSD of about 4-5 A, whereas the Gen7-human complex was very unstable with a variation of RMSD of up to 15 A.
Discussion
The results indicate that deep learning and bioinformatics have the potential to be used in designing highly selective therapeutic agents. This will address the serious problem of off-target toxicity that has hampered the clinical development of earlier HSP90 inhibitors, offering a feasible solution to developing more effective and safer antifungal agents.
Conclusion
This study successfully designed and computationally validated Gen7, a novel de novo inhibitor that selectively targets HSP90 in Rhizopus species. The research provides strong proof of concept for a new class of targeted antifungal agents, offering a promising avenue for developing innovative treatments against drug-resistant fungal infections like mucormycosis.
1. INTRODUCTION
Rhizopus (R. oryzae, R. microsporus, and R. stolonifer) species pose a significant health risk, particularly to immunocompromised persons, due to the presence of invasive fungus. These are the fungi that lead to mucormycosis, a fast-spreading and life-threatening disease [1-3]. The management of mucormycosis is limited to a small number of treatment options as the pathogen is intrinsically resistant to most antifungal agents, including triazoles and even echinocandins, and polyenes like amphotericin B may be toxic. The minimal activity of existing treatment emphasizes the necessity of developing new antifungal approaches capable of bypassing resistance mechanisms [1, 4].
Heat Shock Protein 90 (HSP90) is one of the promising targets in the war against antifungal resistance because it is a highly conserved molecular chaperone that is fundamental to the survival, virulence, and adaptive responses of fungal pathogens. Hsp90 plays a role in protein folding, stabilization, and activation of numerous client proteins in essential cellular processes, such as morphogenesis and antifungal resistance [5, 6].
In yeast (such as Candida albicans) and filamentous fungi (such as Aspergillus fumigatus), it has been established that HSP90 facilitates fast acquisition of adaptation to antifungal stress and contributes to both innate and acquired resistance to azoles and echinocandins [7, 8].
Suppression of HSP90, using both genetic approaches to inhibition, as well as drugs, enhances the efficacy of antifungal agents such as echinocandins and azoles and can overcome drug resistance [9, 10]. Moreover, its inhibition also interferes with important stress-response signaling pathways, such as calcineurin and Lysine Deacetylase (KDAC) signaling, which fungi require to withstand antifungal and environmental stresses. This has also been observed in other plant species like Trichophyton rubrum and Fusarium graminearum [5, 11].
The high level of conservation of HSP90 in eukaryotes is one of the greatest challenges [12, 13]. It is difficult to develop inhibitors that do not affect the human version but only the fungus protein because of the high conservation. Human HSP90 inhibition is associated with serious side effects that render these drugs inappropriate in antifungal therapy, especially in compromised and vulnerable patients [14]. As an example, geldanamycin and its analogs, although effective on fungal HSP90, are very toxic in mammals [15]. Research has discovered possible selective inhibitors, including CMLD013075, that are much more selective for Candida albicans HSP90 by uncovering structural and genomic distinctions among the fungal and human HSP90 [16]. In Cryptococcus neoformans, HSP90 is located on the cell surface and is associated with echinocandin resistance, and inhibition of the protein is synergistic with azole treatment [17].
When HSP90 is inhibited, trichophyton rubrum is more susceptible to itraconazole and micafungin; it leads to decreased colonization of human nails [18, 19]. Aspergillus terreus is sensitive to the HSP90 inhibition and thus reduces its resistance to echinocandins [20, 21]. Although the Saccharomyces cerevisiae has yielded information on the HSP90 functions, Paracoccidioides lutzii and Paracoccidioides brasiliensis exhibit disturbed morphological transitions when HSP90 is inhibited [22, 23]. Equally, in Candida glabrata, the efficacy of echinocandins is increased by the inhibition of HSP90 [24].
HSP90 inhibitors do not have inherent fungicidal properties, yet they can be used in conjunction with the available antifungal agents to make them more effective, especially against drug-resistant fungi. Combination therapy is required because of the various resistance mechanisms of the different fungal pathogens. Nevertheless, such a method adds more complexity to the treatment and the probability of side effects [11]. Although preclinical trials have yielded positive outcomes, clinical trials of HSP90 inhibitors have been difficult due to toxicity and reduced efficacy from off-target effects. In the year 2024, pimitespib was the only HSP90 inhibitor to be approved in Japan for the treatment of gastrointestinal stromal tumors [25].
There are fundamental gaps in the research that hamper the clinical translation of HSP90 inhibitors as antifungals. The first problem is that there are no Rhizopus-specific inhibitors, since attention has been paid to pathogens such as Candida and Aspergillus. Moreover, structural conservation between fungal and human HSP90 is high, which makes it difficult to obtain good selectivity of the inhibitors and avoid off-target toxicity. This has necessitated an urgent requirement to develop new drug design technologies that can take advantage of minute structural differences to result in fungal-specific inhibition.
Hence, the research objective is to fill these gaps by introducing a de novo design method based on deep learning to design a novel protein inhibitor targeting one of the conserved regions in Rhizopus HSP90. Our goal is to find an inhibitor computationally and experimentally that will exhibit a high-quality, steady binding to fungal HSP90 and have minimal interactions with the human homolog. This paper aims to reach a proof-of-concept, generating a novel class of selective antifungal agents that can circumvent the main drawbacks of current therapies through a fusion of conservation analysis, molecular docking, and molecular dynamics simulations.
2. METHODS
2.1. Protein Sequence and Domain Retrieval
The protein sequences for HSP90 were retrieved for all available strains of R. oryzae, R. microsporus, and R. stolonifer from the UniProt database (given in Supplementary File 1) (https://www.uniprot.org/), providing a freely accessible set of protein sequences annotated with functional information [26].
2.2. Conserved Region Identification Using Multiple Sequence Alignment
To investigate the evolutionary trends of HSP90 among Rhizopus across several strains, a protein Multiple Sequence Alignment (MSA) was performed using the TCoffee webserver with default parameters (available at https://tcoffee.crg.eu/) [27]. Protein sequences retrieved previously were used as input to obtain a conserved pattern among the sequences. A custom Python script was generated to extract the most conserved region in the MSA. Additionally, the protein regions of this conserved region for each species of Rhizopus were then used to generate a consensus sequence using a custom script [28].
2.3. Deep Learning Based Protein Inhibitor Designing and Selection of De Novo Proteins
Using the conserved consensus protein sequence among Rhizopus species, Protein Generator (https://huggingface.co/spaces/merle/PROTEIN_GENERATOR) was employed to generate ten different de novo protein sequences and their subsequent structures. A DDPM-style diffusion model was implemented over protein sequence space to generate protein sequence-structure pairs. Starting with RoseTTAFold, the model was finetuned to predict both sequence and structure from partially noised sequences [29]. Training applied losses to both predicted sequence and structure, enforcing the generation of coherent pairs. Diffusion in sequence space enabled the incorporation of potentials to guide the process toward specific properties such as amino acid composition and net charge. Additionally, the model can sample proteins from specific families or use a sequence-to-function classifier to guide generation toward desired sequences.
However, except for the first generated sequence, the remaining generated sequences were manually modified on strategic residue locations to include more hydrogen bond interactions, inducing residues. This approach involved strategically replacing certain amino acids with others that are more likely to form hydrogen bonds, such as serine, threonine, tyrosine, asparagine, and glutamine, whereby the rationale is that a greater number of hydrogen bonds and salt bridges at the interaction interface leads to a more stable and potent inhibitor complex. The 3D structure of these de novo sequences was then predicted using ColabFold (based on AlphaFold), and the top-ranked structures were selected for further analysis based on the pLDDT (predicted Local Distance Difference Test) score [30]. The pLDDT score is a metric used to assess the confidence or quality of structural models predicted by AlphaFold, particularly in predicting the accuracy of specific regions of a protein. This score ranges from 0 to 100, provides an estimate of the confidence in the local structure prediction, with scores above 90 typically indicating high accuracy for both backbone and side chains, scores between 70 and 90 suggesting a largely correct backbone, while regions with pLDDT below 50 may be intrinsically disordered or poorly predicted. For the prediction parameters, num_relax was set to 0, meaning no AMBER relaxation was performed. The template_mode was “none”, indicating no structural templates were used. The MSA options included msa_mode set to “mmseqs2_uniref_env”, pair_mode set to “unpaired_paired”. In the advanced settings, model_type was “auto”; when given a monomer input, this would default to alphafold2_ptm. The num_recycles was set to “3”, and recycle_early_stop_tolerance was “auto” (which would default to 0.0 for a monomer with model_type=”auto”). The relax_max_iterations was 200 (though not applicable here as num_relax was 0), and pairing_strategy was “greedy”. The option calc_extra_ptm was unchecked, meaning it was false. For sample settings, max_msa was “auto”, num_seeds was 1, and use_dropout was unchecked (false).
Molecular dynamics simulations were performed to assess the stability of de novo predicted protein structures using Maestro 12.0 (Schrödinger, LLC). The Protein Preparation Wizard was employed for preprocessing, optimization, and water molecule removal. Salt ions were added to the docked complex, and the SPC force field was chosen as the solvent model. A 50-nanosecond (ns) simulation was run at 300 K. Trajectories were analyzed using the interaction diagram module to calculate Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuations (RMSF). The top three most stable de novo structures were selected based on these results.
2.4. Protein-Protein Molecular Docking Between HSP90 and Designed Inhibitors
The 3D structures of the biggest HSP90 sequences of R. microsporus (A0A0A1PIV1), R. stolonifer (A0A367ILT8) were retrieved from AlphaFold due to the unavailability of the structures on the Protein Data Bank (PDB) (https://www.rcsb.org/) database. While for R. oryzae, the structure was predicted through ColabFold (AlphaFold) due to the unavailability of a pre-predicted structure in the AlphaFold database (https://alphafold.ebi.ac.uk/).
The structures were docked against the top three most stable de novo models obtained in the previous step. For protein-protein molecular docking, the GRAMM (Global RAnge Molecular Matching) algorithm (available at https://gramm.compbio.ku.edu/) was used. GRAMM systematically explores the intermolecular energy landscape, predicting various docking poses that correspond to both stable interactions (deep energy minima) and transient interactions (shallow minima) [31]. Only the top-ranked complexes were selected for further analysis.
Among these complexes, the de novo structure with the best average binding affinity across all three species was chosen for additional investigation. The resulting docking conformations were analyzed for potential protein-protein interactions based on their energy profiles using the PDBSum tool (https://www.ebi.ac.uk/thornton-srv/databases /pdbsum/), which provided detailed visualizations and insights into key interaction features, such as hydrogen bonds, salt bridges, and hydrophobic contacts between the docked proteins [32]. Additionally, as a control, the human HSP90 protein structure was retrieved from AlphaFold and docked with the best-binding de novo structure obtained previously using GRAMM. The interactions were then analyzed using PDBSum to assess differences in hydrogen bonding and other interactions. ProtScale (https://web. expasy.org/protscale/) was used to assess the hydrophobic and hydrophilic regions of the binding site.
2.5. Molecular Dynamics Simulations
The best-binding de novo structure-HSP90 complexes obtained previously were simulated for 30 ns using the same methodology mentioned above. This was conducted to assess the inhibition capabilities of the de novo generated structures against the HSP90 compared to the human control. Furthermore, we assessed the conformational stability of the de novo-HSP90-stolonifer protein in relation to Geldanamycin, a naturally sourced antitumor agent. This benzoquinone ansamycin, synthesized by the bacterium Streptomyces hygroscopicus, serves as a specific inhibitor of HSP90 function through its interaction with the ATP-binding domain of HSP90.
3. RESULTS
3.1. Diversity of HSP90 Protein Lengths in Rhizopus Strains
A total of 12 protein sequences for HSP90 were retrieved from UniProt for various strains of R. oryzae, R. microsporus, and R. stolonifer. R. microsporus is represented by multiple strains, including R. microsporus var. microsporus and R. microsporus ATCC 52813, with sequence lengths ranging from 699 to 738 amino acids. The shortest sequence (304 amino acids) comes from R. oryzae (also known as R. arrhizus var. delemar), a mucormycosis-causing agent, while the longest sequences are from R. microsporus at 738 amino acids. Additionally, R. stolonifer, commonly known as bread mold, has HSP90 sequences of 454, 504, and 699 amino acids, reflecting the variety within this species. Supplementary Document 1 provides information regarding the UniProt ID and strain.
3.2. Identification of Conserved Regions in HSP90 Protein among Rhizopus Strains
To investigate evolutionary trends and identify conserved regions in the HSP90 protein among Rhizopus strains, an MSA was performed using T-Coffee. The analysis yielded intriguing results, revealing a conserved region comprising only 96 residues across the selected strains. This conserved region was extracted using a custom Python script, which was subsequently used to generate a consensus sequence. The conserved region within the MSA is depicted in Fig. (1A), while the extracted sequences are provided in Supplementary File 1. The resulting consensus sequence is: “MFQEIAEDNKXXFDKFYEAFSKNLKLGIHEDTQNRXKLAXLLRYXSTKSGTDXXTSXKDYVTRMPEKQKNIYYITGESRXAXEXSPFLEGFKKKGIEV”, where “X” represents any residue. For R. microsporus, the conserved residues are between 395 and 490, for R. oryzae, it is between 1 and 96, while for R. stolonifer, it is between 360 and 454. Structural visualization of the conserved regions is given in Fig. (1B).
3.3. Structural and Dynamic Analysis of De Novo Protein Structures Reveals Stable Structures
Using the conserved consensus protein sequence among Rhizopus species, the Protein Generator tool (available at Hugging Face) was utilized to generate ten distinct de novo protein sequences, along with their corresponding structural models. The detailed sequences can be found in Supplementary File 1, while the amino acid compositions of these generated sequences are illustrated in Fig. (1C).
The comparison of the amino acid compositions in the gen 1-4 and gen 6-10 indicates strong consistency in the amino acid profiles, indicating that the biochemical environment was stable. However, sequence gen5 has a different amino acid composition in specifics, such as the increased proportion of aspartic acid (D) and glutamic acid (E), which suggests the possibility of a functional or structural ambiguity. In all sequences, some amino acids have the same recurrence rate: Alanine (A), cysteine (C), phenylalanine (F), glycine (G), and isoleucine (I) are represented at percentages of 5.1, 10, 7.1, 5.1, and 1, respectively. On the same note, lysine (K), leucine (L), and methionine (M) are always found at 13%, 7.1, and 2, respectively. Other amino acids, such as asparagine (N), proline (P), glutamine (Q), arginine (R), threonine (T), valine (V), and tyrosine (Y) have also had fixed percentages across the sequences, and they were 2, 6.1, 2, 3.1, 4.1, 6.1, and 2, respectively. It is important to note that serine (S), threonine (T), and tyrosine (Y) have hydroxyl (-OH) groups interacting greatly in hydrogen bonding, and are found in similar levels of 4.1%, 4.1, and 2. Moreover, asparagine (N) and glutamine (Q) contain amide (-CONH2) groups, which allow forming additional hydrogen bonds; glutamine, in its turn, is slightly increased in gen5.
The structures generated were screened based on the pLDDT score, which showed high-quality predicted structures, as shown in Fig. (1D). Additionally, the resulting structures are visualized in Fig. (1E) as superimposed structures. We also used the Maestro 12.0 in order to simulate the three most stable de novo structures. The RMSD and RMSF values analysis shows different trends of protein structural behaviors during a 20 ns simulation, and it offers information on the dynamic stability of different protein structures and their flexibility. The general increase in RMSD is observed in most structures, implying that they have a deviation that increases with the course of the simulation in relation to the reference structure. Interestingly enough, the values of RMSD in Hydro2 are the lowest in the whole simulation, which means that this chemical is the structurally closest to the reference. Conversely, structures, such as 6 and 7, show significant increments of RMSD, especially in the initial few nanoseconds. As an illustration, structure 7 has a steep increase in RMSD in the initial 2 ns, followed by a slow stabilization at the 12 ns point, which indicates that it undergoes significant conformational changes during the first few nanoseconds, then attains relative stability, as seen in Fig. (2B).
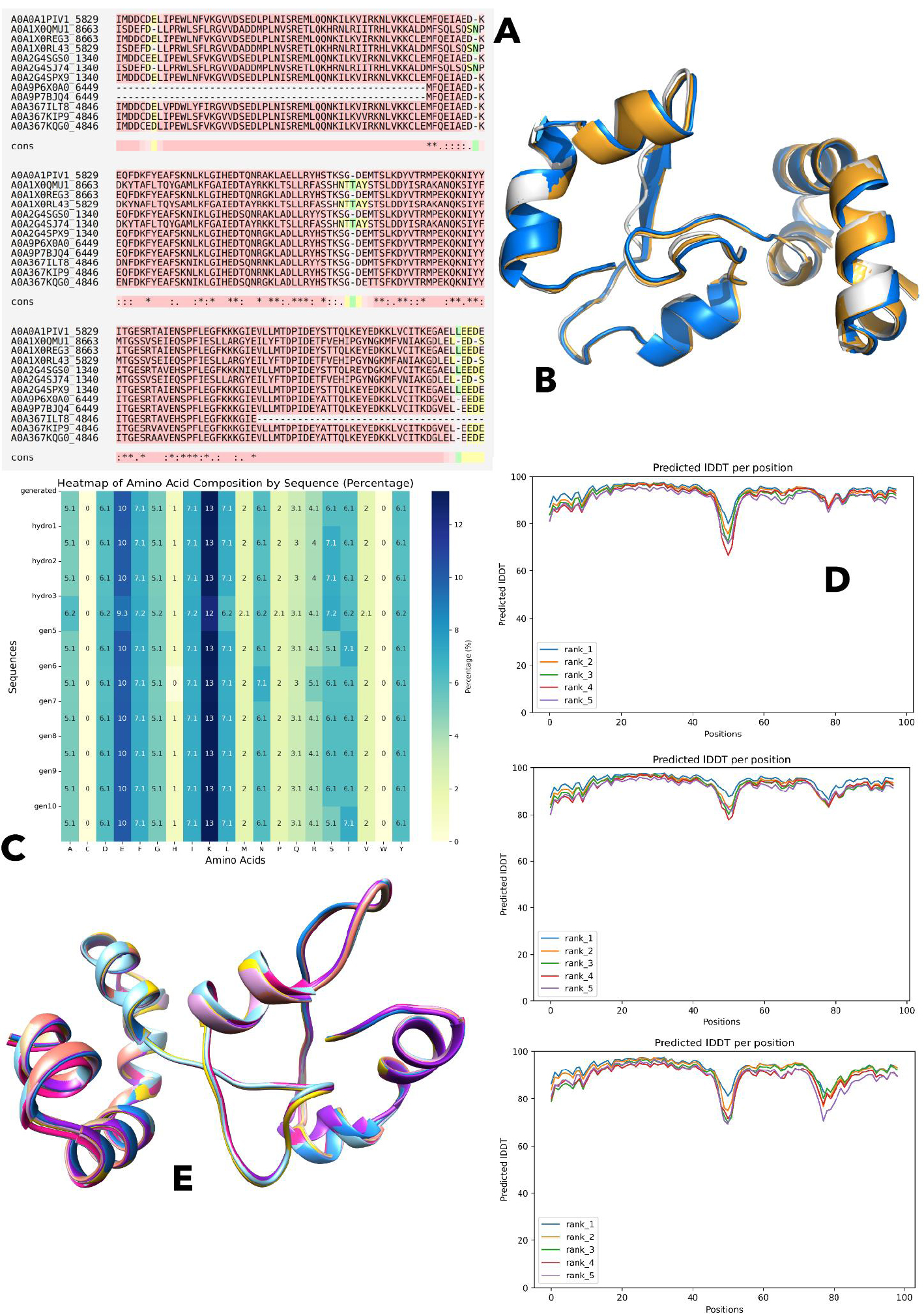
(A) Multiple Sequence Alignment (MSA) highlighting the conserved region.
(B) Structural visualization of the conserved regions identified in the MSA.
(C) Amino acid composition of the ten generated de novo sequences.
(D) Predicted high-quality structures selected based on pLDDT scores.
(E) Superimposition of the generated protein structures to assess structural consistency.
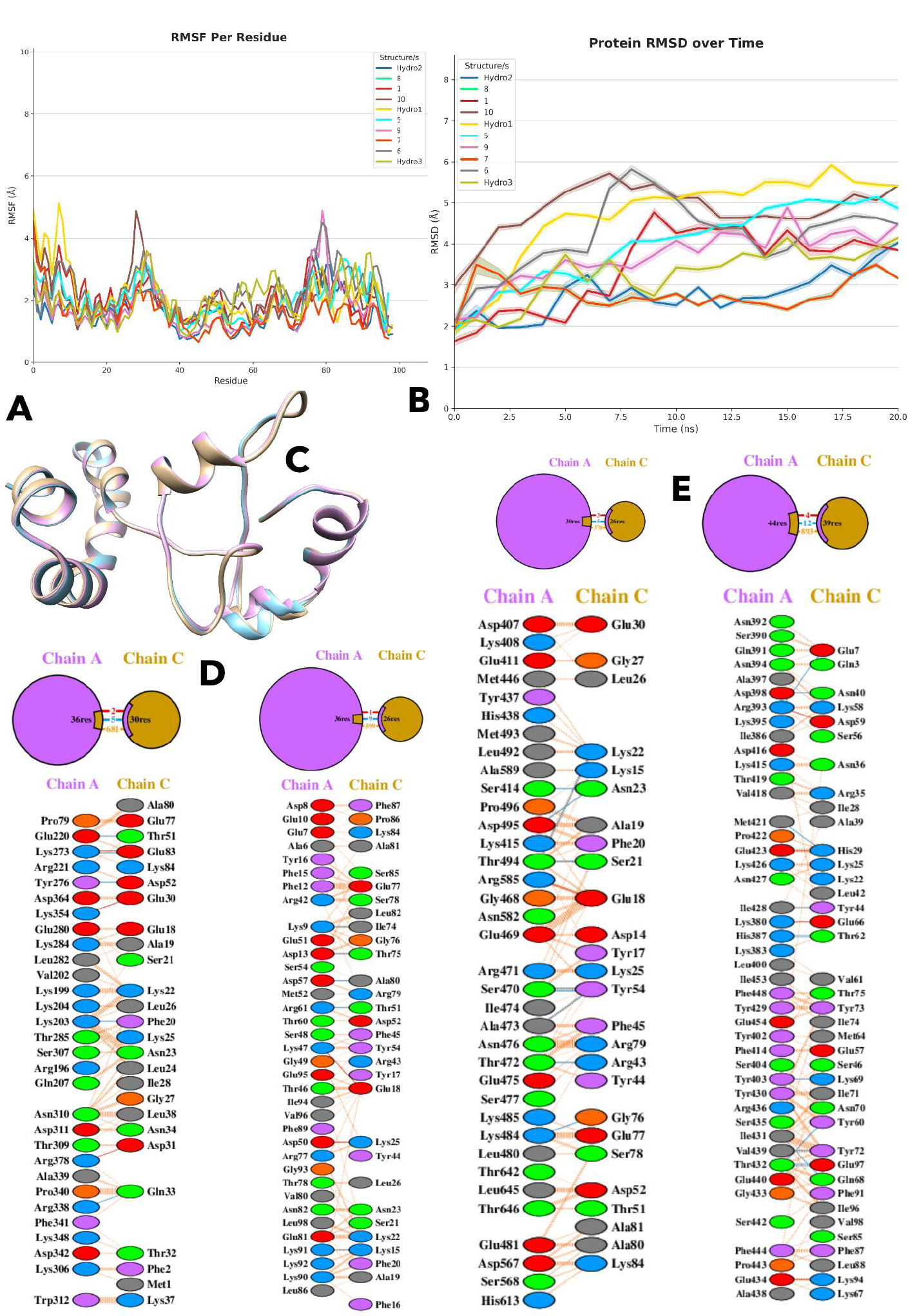
(A) Root Mean Square Fluctuation (RMSF) analysis showing per-residue flexibility of the generated protein structures.
(B) Root Mean Square Deviation (RMSD) plot indicating that structure 7 undergoes early conformational changes before stabilizing.
(C) Superimposed structures of Hydro 2, Hydro 3, and structure 7, identified as the most stable during molecular dynamics simulations.
(D) Hydrogen bonds and salt bridges formed between the de novo designed protein Gen7 and HSP90 from various fungal species and humans, indicating interaction specificity.
(E) Number of interface residues involved in binding between Gen7 and HSP90 from different fungal species and human, reflecting interaction surface size.
Hydro1 and structure 5, in their turn, show the first increase in the RMSD, and then a flat or even decreasing trend, which means that these structures can develop a relatively stable conformation following the initial fluctuations. Notably, structure 8 has variable values of RMSD, indicating that it is sampling a variation of conformations throughout the simulation.
At 10 ns, Hydro2 is also apparent with the lowest RMSD values, whereas structure 6 has the highest RMSD, which is a significant deviation from the reference structure. Additionally, the fact that the values of Hydro2 are low and steady in every part of the simulation is also a representative of the overall stability. By contrast, the RMSD values of Hydro3 are usually within the range of middle values, according to the moderate structural deviations over time. On the whole, structure 7 has a range of 2.8 to 3.5 AA, hydro 2 is between 2.0 and 4.1AA and hydro 3 is between 2.1 and 4.1AA in relation to 3.1 to 5.9 AA on average in other structures.
Moreover, the RMSFD analysis gives an extra view into the versatility of these protein structures per-residue scale, which is presented in Fig. (2A). The values of RMSF of Hydro2 are low in most residues, meaning it has a very rigid structure with only minor changes in flexibility at 25 and 80 residues. However, in contrast, Hydro3 depicts a more diverse RMSF profile and has both areas of flexibility and rigidity. The presence of a significant peak at residue 80, with an approximate 3.5 A, implies that this area was more flexible. This variation indicates that the residues surrounding residue 80 in Hydro 3 undergo a large movement as compared to the less flexible Hydro 2. Structure 7 particularly has large values of RMSF in its beginning, and is then comparatively low when compared with those of its residues throughout the structure.
The flexibility between Hydro 2 and Hydro 3 is also compared, highlighting the overall rigidity of Hydro 2. Hydro 2 is noted to have lower values of RMSF, usually ranging between 1 A and 2 A, implying that there are minimal fluctuations and the structure is more stable. However, the Hydro 3 values of RMSF are 0.8 to 3.5 A, which is more dynamic. The greatest variation occurs in structure 10, where there is a maximum RMSF of about 5 A around residue 25, such that this area is the most flexible in all the analyzed structures. These differences in flexibility and rigidity point to the difference in structural behaviors with Hydro 2, Hydro 3, and structure 7 becoming the most stable, which were shortlisted as structural protein-protein docking with HSP90. These three structures are visualized as superimposed structures in Fig. (2C).
3.4. Designing of Fungal-Specific HSP90 De Novo Protein Inhibitor
The 3D structures of HSP90 of R. microsporus, R. stolonifer, and R. oryzae were docked against the gen7, hydro2, and hydro3 (the three most stable de novo models). For protein-protein molecular docking, the GRAMM was used by specifying the highly conserved regions of each protein structure (as mentioned above).
The molecular docking results revealed that for the Gen7 structure, R. oryzae exhibited the strongest binding affinity with a score of -494, followed by R. microsporus at -452 and R. stolonifer at -422. The average docking score for Gen7 was calculated as -456, indicating that, on average, the ligands designed using this method demonstrated relatively strong binding across all species tested. However, the human receptor also showed a relatively strong interaction with a score of -443, but it is still below the fungal species’ average binding score.
The Hydro 2 structure showed a slightly different trend. R. oryzae displayed a weaker binding affinity than with the Gen7 structure, with a docking score of -416, while R. microsporus remained relatively consistent with a score of -455. Stolonifer, however, had a slightly stronger interaction than with Gen7, with a docking score of -433. The average score for Hydro2 was -434, slightly worse than the Gen7 structure for these species.
With the Hydro 3 structure, R. oryzae had the weakest binding among the methods, with a score of -443. R. stolonifer and R. microsporus showed improved binding affinities relative to Hydro 2, with scores of -430 and -439, respectively. The overall average score for Hydro 3 was -437, indicating that this method produced results between those of Gen7 and Hydro 2, with moderate binding affinities across the species tested. Among these complexes, the Gen7 structure with the best average binding affinity of -456 was chosen for further investigation.
The resulting docking conformations were analyzed for potential protein-protein interactions based on their energy profiles using the PDBSum tool, which provided detailed visualizations and insights into key interaction features, such as hydrogen bonds, salt bridges, and hydrophobic contacts between the docked complexes. The docking results indicate that the de novo designed protein Gen7 interacts with different fungal species with varying effectiveness.
Among the species, the number of interface residues, which reflects the size of the binding interaction, is largest for R. stolonifer (44 and 39 residues) and smallest for the human protein (30 and 26 residues from de novo structure), suggesting a less extensive interaction in humans, given in Fig. (2E). R. microsporus and R. oryzae have intermediate interface residue counts, with 36 and 26 for R. microsporus and 36 and 30 for R. oryzae. Similarly, R. stolonifer also has the highest number of stabilizing hydrogen bonds (12), whereas the human protein has only 5. R. microsporus forms 9 hydrogen bonds, and R. oryzae forms 5, indicating varying degrees of stability. Although the human protein has a relatively higher number of salt bridges (3), R. stolonifer has 4, R. oryzae has 2, and R. microsporus only has 1, as shown in Fig. (2D).
However, the human protein's low non-bonded contacts (370, the lowest among the species) further suggest a weaker overall interaction. In contrast, R. stolonifer has 893 non-bonded contacts, while R. microsporus and R. oryzae have 599 and 681, respectively, highlighting more extensive interaction networks in these species. While Gen7 shows moderate binding with the human target, the weaker hydrogen bonding, smaller interface, and fewer contacts suggest that it may not be as effective for human applications compared to its performance with R. oryzae or R. microsporus.
Through a detailed investigation of hydrogen bond interaction, it was observed that in the interaction between Gen 7 and human HSP90, hydrogen bonds are less likely to occur at highly conserved regions. The residues involved include LYS 203, GLU 220, LYS 273, TYR 276, and ARG 338 on Chain A, binding with PHE 20, THR 51, GLU 83, ASP 52, and GLN 33 on Chain C. The distances range from 1.75 Å to 3.33 Å, suggesting moderate to strong interactions. The key takeaway here is that these residues are not from highly conserved regions of HSP90 identified and used for generating the de novo structures in this study, indicating that Gen 7 avoids binding to critical functional regions in humans.
In R. oryzae, Gen 7 shows stronger and more precise hydrogen bonding interactions. The residues involved, such as LYS 9, ASP 13, and ARG 61 on Chain A, bind to ILE 74, THR 75, ALA 80, and THR 51 on Chain C with distances ranging from 2.03 Å to 3.14 Å. Notably, these residues, especially the LYS-ILE and ASP-THR interactions, occur at highly conserved regions of R. oryzae HSP90. The tight bonding distances reflect a more stable interaction. The protein-protein docked R. oryzae-gen7 is given in Fig. (3C).
Moreover, in R. microsporous, a similar pattern of interaction is observed, with Gen 7 forming hydrogen bonds with conserved regions of HSP90. The residues SER 414, SER 470, THR 472, and LYS 484 on Chain A form bonds with ASN 23, TYR 54, ARG 43, and GLY 76 on Chain C, with distances ranging from 2.55 Å to 3.30 Å. The critical interaction between THR 472 and ARG 43 (distance: 2.58 Å) as well as SER 470 and TYR 54 (distance: 2.70 Å) indicates strong binding to highly conserved residues. The protein-protein docked R. microsporous-gen7 is given in Fig. (3A).
Finally, in R. stolonifer, Gen 7 forms numerous hydrogen bonds, such as between HIS 387, ARG 393, ASP 398, and ILE 428 on Chain A, and THR 62, GLU 7, ASN 40, and TYR 44 on Chain C. Distances range from 2.08 Å to 3.23 Å. Importantly, many of these bonds, like the THR 432 to GLU 97 interaction (distance: 2.08 Å), target conserved areas of the R. stolonifer. The protein-protein docked R. stolonifer-gen7 is given in Fig. (3B).
3.5. Molecular Dynamics Trends of Designed Inhibitor with Fungal and Human HSP90
The best-binding gen7-HSP90 complexes obtained previously were simulated for 30 ns using DESMOND, aiming to assess the inhibitory capabilities of gen7 against HSP90 compared to the human control. The RMSD analysis of the four protein structures over the 30 ns molecular dynamics simulation reveals distinct patterns for each complex. The gen7-microsporus complex shows a general upward trend in RMSD over time, with some minor fluctuations. Notably, there is a significant increase in RMSD between 5 ns and 10 ns, after which the structure enters a period of relative stability, maintaining an RMSD range of 8-10 Å. This indicates that, despite the initial rise, the structure stabilizes as the simulation progresses. Compared to the other complexes, gen7-microsporus exhibits a relatively strong stability throughout the simulation. However, the gen7-stolonifer complex also stabilizes after reaching equilibrium around the same time as gen7-microsporus. It achieves a stable RMSD range between 4-5 Å, showing stronger stability than gen7-microsporus. However, a notable peak of 6.1 Å is observed at 22 ns, after which the RMSD returns to its stable range of 4-5 Å, reflecting minor perturbations but overall consistent stability, as given in Fig. (3D).
In contrast, the gen7-oryzae complex exhibits a pronounced upward trend in RMSD with more significant fluctuations than both gen7-microsporus and gen7-stolonifer. Large peaks are observed at 5 ns, 8 ns, 15 ns, and 23 ns, with the RMSD values consistently fluctuating between 7.5 Å and 12 Å throughout the simulation period. This pattern indicates a more dynamic and less stable complex compared to the previous structures.
Similarly, the gen7-human complex displays substantial fluctuations, showing a similar trend to that of gen7-oryzae, where the structure fluctuates significantly throughout the entire simulation period. RMSD values range from 8 Å to 15 Å, with a continuous upward trend and no stability observed at any point in the simulation. Large peaks are particularly pronounced after 20 ns, with the highest peak at 25 ns reaching 14.1 Å, further highlighting the lack of stability in this complex compared to the others. Of note, gen7-stolonifer demonstrates the strongest stability, followed by gen7-microsporus, while gen7-oryzae and gen7-human exhibit significant fluctuations and less stable behavior over the 30 ns simulation period.
RMSF analysis reveals distinct fluctuations across the structures of gen7 variants. Both gen7-human and gen7-microsporous exhibit relatively high RMSF values (~20 Å) at the starting residues, whereas gen7-stolonifer and gen7-oryzae display lower values (~5 Å) in the same region. All structures remain stable up to residue 200, with gen7-stolonifer showing a spike up to 14 Å, while gen7-oryzae has the smallest peak (~5 Å) among the variants. Around residue 225, gen7-microsporous shows a notable peak, and gen7-human has several smaller peaks in this region. Gen7-oryzae displays the largest peak (~20 Å) at residue 300. After this, the structures generally remain stable, though gen7-human and gen7-microsporous experience high RMSF peaks (~32 Å and ~23 Å, respectively) at residues 700 and 690, before returning to stable values around 5 Å, as given in Fig. (3E).
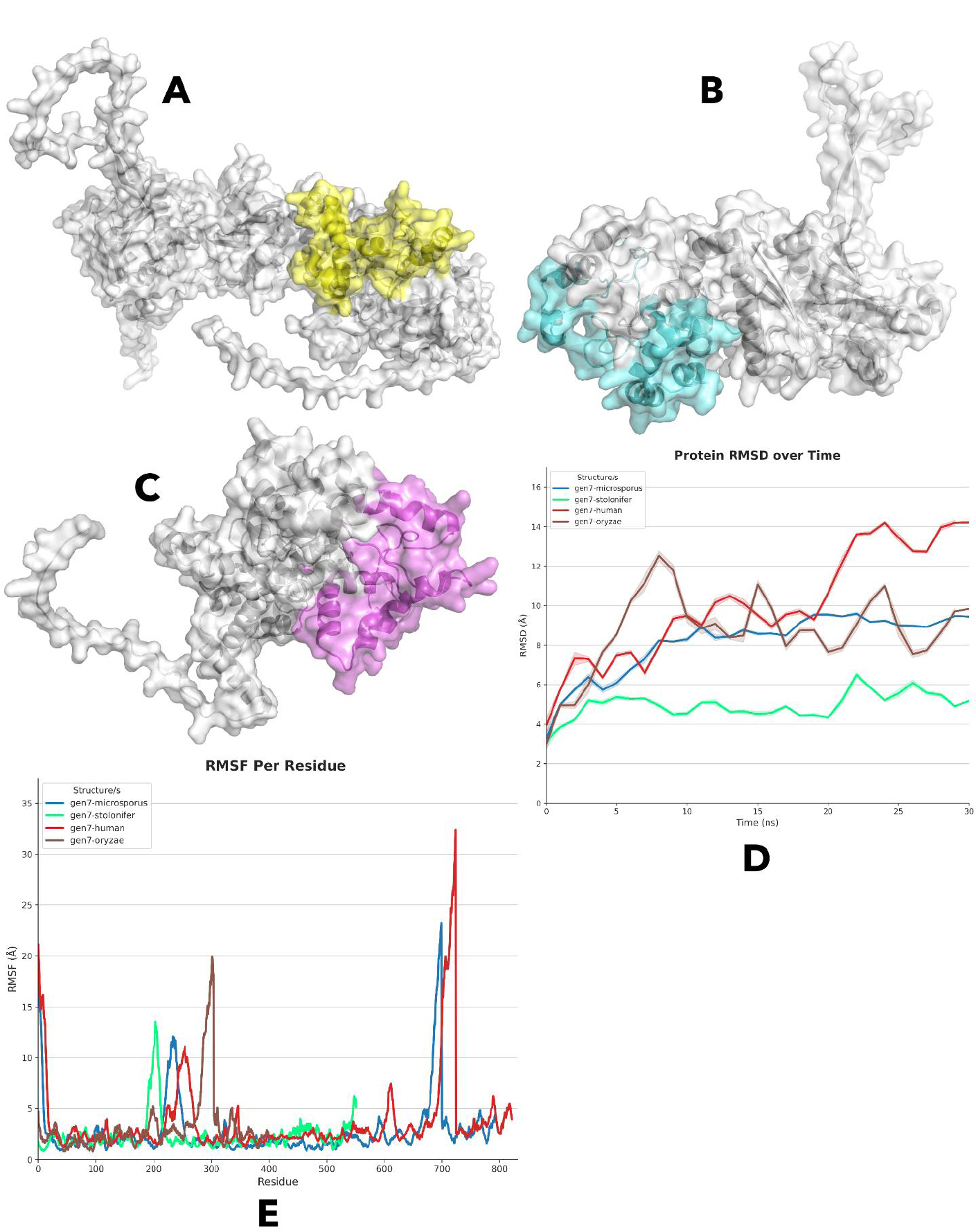
(A-C) Protein-protein docked complexes of Gen7 with HSP90 from R. microsporus, R. stolonifer, and R. oryzae, respectively.
(D) RMSD analysis over a 30 ns simulation showing distinct structural dynamics of each Gen7-HSP90 complex, including human.
(E) RMSF analysis highlighting fluctuation patterns across Gen7 complexes with fungal and human HSP90.
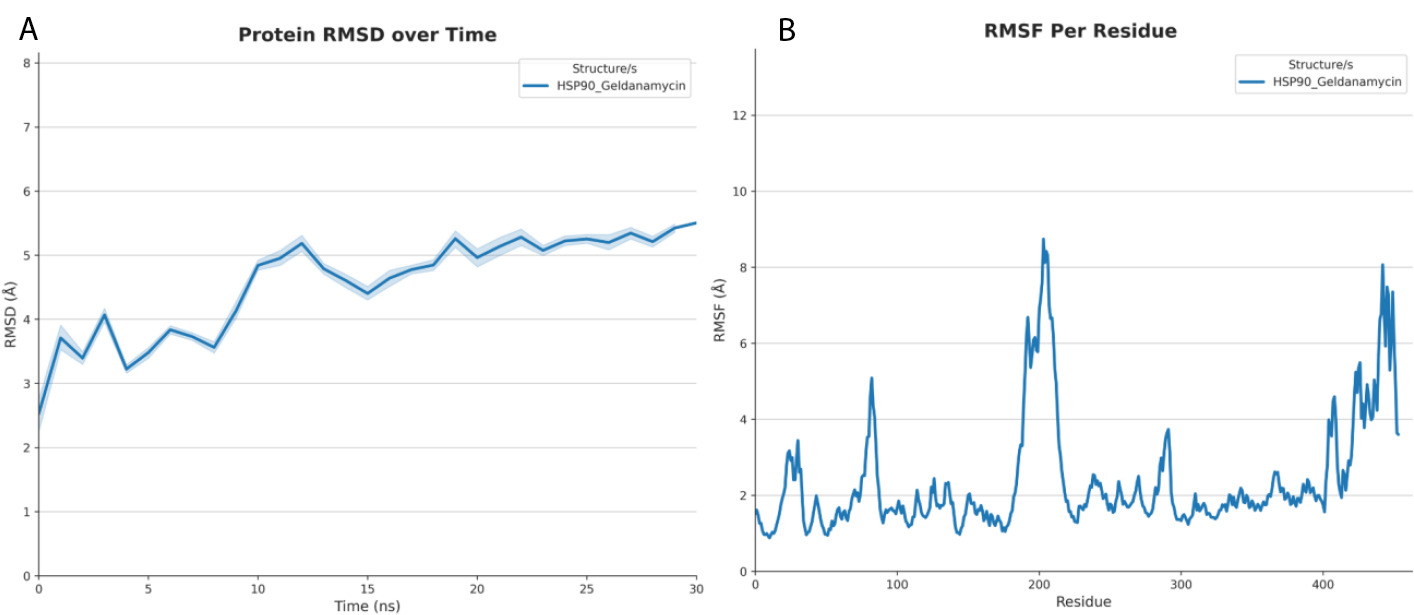
(A) RMSD analysis of the HSP90-Geldanamycin complex over time.
(B) RMSF analysis showing residue-level flexibility in the HSP90-Geldanamycin complex.
Finally, based on the molecular dynamics analysis of the HSP90-Geldanamycin complex, we compared it with gen7-stolonifer, highlighting differences in stability and flexibility. The HSP90-Geldanamycin complex exhibits an initial rise in RMSD followed by a sharp conformational change between 7 and 11 ns, after which it stabilizes around 5–5.5 Å. In contrast, the gen7-stolonifer complex stabilizes more quickly, maintaining an RMSD of approximately 4.5 and 5.5 Å throughout most of the simulation and ending near 5 Å, without the pronounced shift seen with Geldanamycin. This suggests that gen7-stolonifer promotes a more consistent and earlier stabilization. Regarding residue flexibility (RMSF), the Geldanamycin-bound complex shows multiple flexible regions, particularly at the N-terminus, around residues 90–100, and significantly in the C-terminal region (residues ~400–460), with a peak around 200–210. In comparison, gen7-stolonifer generally induces lower overall fluctuations, especially reducing flexibility in the C-terminal region and lacking the 90–100 peak seen with Geldanamycin. However, it causes a higher degree of flexibility in the 200–220 region than Geldanamycin. Taken together, gen7-stolonifer appears to stabilize the protein earlier and more smoothly than Geldanamycin, while altering the flexibility profile by decreasing motion in some regions (e.g., the C-terminus) and increasing it in others (notably around residues 200–220). Therefore, we show here a potentially more stable inhibitory mechanism against HSP90 than geldanamycin. The RMSD and RMSF for HSP90-Geldanamycin are visualized in Fig. (4A-B), respectively. Finally, we summarize the findings in Table 1.
| Target Protein | Docking Analysis (Binding and Specificity) | MD Simulation (Stability) |
|---|---|---|
| R. stolonifer HSP90 | • Strongest Interaction • 12 Hydrogen Bonds • 4 Salt Bridges |
• Most Stable Complex • Stable RMSD (4-5 Å) • More stable than Geldanamycin complex |
| R. microsporus HSP90 | • Strong Interaction • 9 Hydrogen Bonds • 1 Salt Bridge |
• Stable Complex • Stable RMSD (8-10 Å) after initial rise |
| R. oryzae HSP90 | • Good Interaction • 5 Hydrogen Bonds • 2 Salt Bridges |
• Less Stable • High fluctuations in RMSD (7.5-12 Å) |
| Human HSP90 | • Weakest Interaction • 5 Hydrogen Bonds • 3 Salt Bridges |
• Unstable Complex • Significant fluctuations; no stability observed (RMSD up to 15 Å) |
3.6. Hydropathic Characterization of the HSP90 Binding Pocket
To further guide the design of new drug candidates, an analysis of the HSP90 binding site was conducted to map its hydrophilic and hydrophobic characteristics using ProtScale. The results indicate that the conserved binding pocket across Rhizopus species is predominantly hydrophilic, a finding consistent with our docking analysis that identified an extensive network of hydrogen bonds and salt bridges stabilizing the Gen7-HSP90 complex. Specifically, in R. stolonifer, which demonstrated the strongest interaction stability, the binding interface (residues 360-454) exhibits high hydration potential, creating a favorable environment for the numerous polar interactions observed. However, this region is not uniform; the analysis also revealed distinct hydrophobic pockets, such as the area around residue 391, suggesting an amphipathic nature. Similarly, the binding regions in R. microsporus (residues 395-490) and R. oryzae (residues 1-96) are also largely hydrophilic but interspersed with hydrophobic patches. This detailed mapping underscores the need for optimal inhibitor design to consider this amphipathic topology. Future drug candidates could be enhanced by incorporating both polar functional groups to engage with the hydrophilic surface and nonpolar moieties strategically positioned to occupy the hydrophobic pockets, thereby maximizing binding affinity and specificity.
4. DISCUSSION
Developing fungal-specific HSP90 inhibitors is difficult because of the high conservation between fungal and human HSP90, leading to off-target toxicity and limited therapeutic efficacy. This study utilized a novel deep learning approach to design de novo inhibitors that specifically target HSP90 in clinically significant Rhizopus species. The aim is to improve inhibitor specificity and efficacy while minimizing side effects, leveraging deep learning's successful application in designing therapeutics for deadly diseases such as bacterial infections, cancer, neurodegenerative diseases, and cardiovascular diseases [33, 34, 35, 36, 37].
The findings of this work provide important insights on the diversity, evolutionary conservation, and structural stability of the HSP90 proteins in the various Rhizopus species and their strains, and this indicates that the HSP90 proteins are dynamic in various species and strains. In particular, R. oryzae (synonyms R. arrhizus var. delemar) introduces the shortest ones, whereas R. microsporus has the longest ones. This implies that there may be differences in the functional roles or regulatory needs of HSP90 across these organisms, which may be indicative of differences in the stress response mechanism or differences in the degree of environmental adaptation. These differences could also be attributed to differences in the pathogenicity of strains. The conservation mechanism of HSP90 in fungi has been reported in other studies in the past [13, 38, 39]. The multiple MSA has indicated the uneven distribution of the conserved areas among the species, with R. microsporus, R. oryzae, and R. stolonifer conserved in various regions of the sequence. This conservation difference might indicate species-specific functions of HSP90 protein, yet which are nonetheless needed by protein-folding core functional domains, as found in previous studies, including an example in which conservation is observed in HSP90 domain structure, including an N-terminal ATPase domain, a middle domain to interact with clients, and a C-terminal domain to dimerize which are required in the functions of HSP90 in protein homeostasis and environmental stress. Additionally, one of the studies found that in Candida albicans, HSP90 is the only protein that regulates virulence functions such as morphogenesis and drug resistance. However, this is not the case with C. albicans, where HSP90 regulates the yeast-to-filamentous transition, which is an important pathogenicity process [8, 41]. It has been demonstrated that non-coding influences can drive these processes, i.e., post-translational modifications, including phosphorylation, which can adversely impact the virulence by changing the stability of the protein and interactions with the client [42]. Fungal and human HSP90 Comparative analysis of fungal and human HSP90 has shown differences in functionality, particularly in their interactions with co-chaperones and in ATPase activity [40, 43].
The sequence and dynamics of these de novo proteins indicate that most of the resulting sequences exhibit variations in biochemical stability. Nevertheless, the unique amino acid structure of sequence Gen5, with high concentrations of aspartic acid and glutamic acid, indicates the possible differences in functionality that could be studied further in the interaction or particular adhesion characteristics. Earlier research has indicated that de novo inhibitors that are designed to have stable backbones and unique amino acid sequences can be better in terms of binding, to enhance their molecular recognition and affinity to the target proteins. Researchers have used fine-tuning models such as RoseTTAFold to denoise design tasks in protein structure, creating generative models capable of solving a wide variety of design tasks, such as protein monomer design, binder design, and symmetric oligomer assembling tasks. The correctness and functionality of these designs are proved by experimental findings; the cryogenic electron microscopy structure of a designed binder is almost similar to the model [44, 45, 46]. Besides, our data obtained on molecular dynamics simulations gave some vital information on the dynamic stability of these designed proteins, and it was found that out of the ten structures created, only three Hydro 2, Gen 7, and Hydro 3 structures were relatively stable compared to others. It is worth noting that Hydro 2 exhibited a high level of stability, and its RMSD and RMSF levels were constantly low throughout the simulation, which indicates that the given structure could be used as a powerful basis for fungal HSP90 inhibition. Gen 7 and Hydro 3 were also stable, but had more fluctuations, potentially indicating that the two are more flexible and adaptable, which, under certain biological conditions, can be beneficial. It is especially applicable in the context of the interaction dynamics of the structures with HSP90 in any given fungal species. Hydro 2 exhibited outstanding stability with low RMSD and RMSF, and this reflects that it can be greatly exploited to inhibit fungal HSP90. The stable Gen 7 and Hydro 3 had more fluctuations, and this is an indication of flexibility that could help in some biological situations, as reported earlier [44, 46]. The molecular docking study between the de novo protein structures and HSP90 of R. microsporus, R. oryzae, and R. stolonifer showed that Gen 7 had the highest average binding affinity with R. oryzae, suggesting it could be a potent inhibitor of this pathogenic species. Interestingly, despite the fact that Gen 7 also showed moderate interaction with human HSP90, the interaction of binding was weak and less dense compared with the fungal species. This selectivity is important in the formulation of therapeutic agents that can act on fungi pathogens with a reduced off-target effect in humans. The step-by-step hydrogen bond and salt bridge interactions found in the docking assays also confirm the hypothesis that Gen7 binds more to conserved regions of fungal HSP90, those that formed the largest interaction network with Gen 7, and that they are R. stolonifer. Salt bridges are also highly useful in drug potency, where the combination of electrostatic attraction and hydrogen bonding is more potent than the simple hydrogen bonds. They usually happen in proteins between the positive Lys or Arg residues and the negative Asp or Glu residues. Salt bridges are important in protein-protein interactions, protein folding, protein recognition, protein conformational rigidity, and protein stability [47]. This paper also demonstrated that the ability of the inhibitor to act was due to the formation of a salt bridge between a positively charged group of the inhibitor and the negatively charged C-terminus of the target enzyme. Replacement of the charged group by a neutral group resulted in a high activity loss, pointing to the need for salt bridges in stabilizing N-myristoyltransferases (NMT)-ligand complexes as well as their possible use as a rational drug design tool. These results were further confirmed by our molecular dynamics simulation, which showed that Gen7 Hsp90 fungi were more stable than Gen7 Hsp90 human. The gen7-R. stolonifer and gen7-R. microsporus complexes had a constant RMSD value across the 30 ns simulation, which denotes the robust and affirmative interaction. However, the gen7-R. oryzae and gen7-human complexes were more variable, implying that the interactions were weak and unstable. Gen 7 selectivity for fungal HSP90 and the less avid binding of the human protein are important advances towards the creation of specific antifungal therapy applications. Recent research has examined the critical importance of MD simulations in understanding the complex interactions between protein stability, dynamics, and functionality. For example, a study of SARS-CoV-2 spike protein variants found to have a higher capacity to stabilize structure, especially the omicron variant, a major factor contributing to greater viral infectivity. Equally, MD simulations of this type, which estimate the binding of the ligand, showed that native forms were more stable than decoys, thus improving structure-based drug design approaches [48, 49, 50, 51, 52]. Thus, those structures that are more stable upon formation of a protein-inhibitor complex are more likely to undergo successful binding and make therapeutic agents.
Nonetheless, this research has a number of weaknesses that must be noted. First, the results are purely computational, and they were not experimentally validated. Its efficacy, stability, and selectivity as predicted by the de novo inhibitor Gen 7 should be verified using in vitro activity and later justified in in vivo models to determine its real (therapeutic) potential. Second, the research used AlphaFold-predicted protein structures in certain species, which, even though highly accurate, might not accurately reflect the native protein structures. Molecular dynamic simulations (30-50 ns) may also not be long enough to include all the long-term conformational dynamics of the protein-inhibitor complexes. These restrictions provide significant opportunities for future research in order to make these computational results a clinically viable antifungal agent. Finally, the paper has identified the applicability of protein design based on bioinformatics in the design of selective antifungal compounds. These studies of evolutionary analysis, structural modeling, molecular dynamics simulations, and docking studies offer a holistic approach to fungal-specific inhibitors identification and optimization. Further optimization of the Gen 7 structure to increase its specificity and efficacy, and in vitro and in vivo testing to confirm its potential as a therapeutic agent, could be considered for future work. The method used in this research can be applied to other pathogenic fungi, thus broadening antifungal drug research and creating new channels of fungal infection control.
CONCLUSION
This paper demonstrates that a deep learning approach can be used to develop selective inhibitors against HSP90 in clinically relevant Rhizopus species. Through the study of evolutionary trends and the design of complex proteins using current protein design tools, we were able to produce the de novo inhibitor Gen 7, which had high binding affinity ability to fungal HSP90 but had little interaction with human proteins. The differences in the lengths and conserved regions of the HSP90 protein among Rhizopus species indicate special functional purposes depending on the evolutionary pressures. Gen 7 proved to be extremely stable and selective, as confirmed by molecular dynamics studies and docking studies, and an augmented salt bridge formation further strengthened it. Moreover, the HSP90-Geldanamycin comparison complex with gen7-stolonifer indicates that gen7-stolonifer may be able to offer a more stable inhibitory mode of action over HSP90 compared to Geldanamycin due to earlier and smoother stabilization and changes in the flexibility profile. These results highlight the possibility of the use of bioinformatics-based protein design to develop specific antifungal agents. The Gen 7 structure needs to be optimized in future studies to be more effective, and in vitro and in vivo analyses should be carried out to determine its therapeutic value. The study forms the basis for developing antifungal agents that can be able to effectively counter the effects of fungi and reduce off-target effects on the human body.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: M.S.F.: Study conception and design; Y.N.H.: Analysis and interpretation of results; S.S.: Investigation. All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| HSP90 | = Heat Shock Protein 90 |
| RMSD | = Root-Mean-Square Deviation |
| MSA | = Multiple Sequence Alignment |
| pLDDT | = Predicted Local Distance Difference Test |
| ns | = Nanosecond |
| RMSF | = Root Mean Square Fluctuations |
| PDB | = Protein Data Bank |
| GRAMM | = Global Range Molecular Matching |
| KDAC | = Lysine Deacetylase |
| MD | = Molecular Dynamics |
| NMT | = N-myristoyltransferases |
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
ACKNOWLEDGEMENTS
Declared none.

