All published articles of this journal are available on ScienceDirect.

Advancements in Vaccine Development: A Comprehensive Design of a Multi-Epitopic Immunodominant Peptide Vaccine Targeting Kyasanur Forest Disease via Reverse Vaccinology
Authors Info & Affiliations
Abstract
Introduction/Objective
Kyasanur Forest Disease (KFD), caused by the Kyasanur Forest Disease Virus (KFDV), is a tick-borne haemorrhagic fever endemic to South India and spreading to neighbouring states. The formalin-inactivated Chick Embryo Fibroblast (CEF) vaccine currently in use provides only short-term protection, requires repeated inoculations, and has limited coverage. A safe, simple-to-administer vaccine, which includes chills, fever, and headaches, was designed as a multi-epitope peptide vaccine (MEPV) against the immunodominant E protein of KFDV by using cutting-edge immunoinformatic and reverse vaccinology approaches.
Methods
Ten KFDV strains (1962–2016) were retrieved from NCBI and screened for antigenicity. The sequence of the E protein of the selected strain was screened for CTL, HTL, and B-cell epitopes using IEDB, NetMHCpan, and ABCPred. Predicted epitopes were evaluated for antigenicity, allergenicity, toxicity, immunogenicity, and conservancy across all the shortlisted strains. Potential epitopes were linked with suitable linkers to form the PKFDVac-I construct. Its physicochemical properties, structure stability, and immunogenic potential were evaluated using Expasy ProtParam, PSIPRED, AlphaFold, molecular docking with TLR-4, molecular dynamics simulation, and C-ImmSim immune simulation.
Results
Sixteen epitopes (5 CTL, 3 HTL, 8 B-cell) cleared all screening criteria and were included in PKFDVac-I, a 279-amino-acid construct with a molecular weight of 29.16 kDa. The vaccine demonstrated high antigenicity, non-toxicity, non-allergenicity, solubility, and stability. Docking was found to be good, with a TLR-4 binding affinity of -1150.78 kcal/mol (Piper energy), supported by 387 non-bonding interactions. A 100-ns molecular dynamics simulation confirmed the stability of the complex. Immune simulation also anticipated robust humoral and cellular immunogenicity, higher antibody titers, long-lived persistence of memory cells, and robust IFN-γ induction.
Discussion
PKFDVac-I had favorable immunological properties in silico. The design comprises conserved epitopes that are antigenic, safe, and immunogenic to the tested Indian KFDV strains from 1962 to 2016, ensuring lineage representativeness. Molecular docking and simulation reveal a stable interaction between receptors, and immune simulations predict durable adaptive immunity.
Conclusion
PKFDVac-I is a proposed multi-epitope peptide vaccine candidate for Kyasanur Forest Disease. The integration of diverse epitopes into a cohesive vaccine prototype demonstrates a promising avenue for custom synthesis and application in immunization strategies. The design represents a significant advancement in the evolution of KFD vaccines and warrants further in vitro and in vivo validation.
1. INTRODUCTION
Kyasanur Forest Disease is an arboviral illness that causes fever and bleeding, spread by arthropods, particularly ticks [1]. Viral diseases account for a considerable burden of global morbidity and mortality, with many being zoonotic in nature, originating in animal hosts before crossing species barriers to infect humans [2]. The zoonotic virus was found to have been distributed in Karnataka, South India (1957) [3]. After a 3–8 day incubation period, KFD symptoms, which include chills, fever, and headaches, appear abruptly. The manifestation of intense myalgia, emesis, gastrointestinal disturbances, and hemorrhagic complications may present three days after the initial onset of symptoms [4]. Case studies on KFD indicated that it was restricted to Karnataka's arboraceous Shimoga district. Recent reports suggest that it has spread to Tamil Nadu (Nilgiris), Kerala (Wayanad & Malappuram), Maharashtra (Sindhudurg), and Goa (Pali) [5]. Between 1957 and 2017, approximately 9,594 cases of Kyasanur Forest Disease (KFD) were reported across various districts along India's western coast. From 1957 to 2020, around 3,314 monkey deaths have been ascribed to KFD [6]. On an annual basis, approximately 160 instances of human cases are documented, exhibiting a CFR of 2.4% [7]. The disease is labeled as seasonal, usually reported from December to May [8]. A study conducted on serosurvey samples from Kingaon-West Bengal (1962), Kutch and Saurashtra-Gujarat (1971), Parbatpur-Rajasthan (1971), and the Andaman and Nicobar Islands (2002) found evidence of hemagglutination inhibition antibodies of KFDV [9]. Chinese researchers have reported similar diseases caused by the Nanjianyin virus [10], Saudi researchers have reported the Alkhurma Hemorrhagic Fever (AHF) since 1995 [11, 12] in the dry season. Individuals who trekked to forests [9] for the purpose of collecting firewood, grasses, and various other forest-derived products were observed to exhibit instances of human cases [13]. The most typical way for ticks to spread the infection to people is by contact with an infected host, particularly one that is either ill or has recently succumbed to illness, such as a monkey [14]. Human-to-human transmission is not yet reported in the case of KFD [15].
Several vaccines were tested to combat the disease, in addition to the currently used formalin-inactivated CEF KFD vaccine [16]. Studies carried out in KFD affected districts of Karnataka during 1990-92 reported an efficacy of 79.3% for one dose of the vaccine, and 93.5% for two doses and in 2005-10, effectiveness was found to be 62.4% in subjects who took two doses and 82.9% in those who took a booster dose after two doses [17, 18]. Some researchers have speculated that the lower efficacy of the vaccine may be due to drifts and diversification from KFDV strains currently circulating compared to the strain used initially to make the vaccine. This indicates a clear need for a detailed study and a highly effective vaccine to contain the spread of the disease [19].
Given that the KFD virus is classified as a Biosafety Level 4 (BSL-4) category pathogen, this study has employed advanced methodologies, including a promising reverse vaccine approach and immuno-informatic tools, to design a multi-epitope peptide vaccine (MEPV) construct aimed at inducing an immune response. MEPV targets specific epitopes of the pathogen and potentially improves efficacy. Vaccines that target multiple epitopes are more successful, as they reduce the likelihood that the pathogen may evolve escape mutants. Since they don't contain entire pathogens — live or attenuated — they are generally safe. Furthermore, these vaccines are highly adaptable, allowing for customization to meet the specific needs of individuals or populations by taking into account genetic variability.
The diameter of the KFD virus is about 25 nm, and the length of the +ve strand RNA genome is nearly 11 bp [20]. It encodes a single polyprotein comprising the structural proteins - E, C, and prM/M and the non-structural proteins (NS-1, NS-2a and b, NS-3, NS-4a and b, and NS-5) after translation [21]. The C protein of KFDV plays two crucial roles in the infection process. It encapsulates viral RNA for protection and also interacts with various host proteins to stimulate virus replication. Nonstructural proteins are proteases that are primarily responsible for cleaving the polyproteins and regulating the host cell response either by directly interacting with the C protein, NS-2a, and NS-3, aiding in virus assembly, or through interactions with structural proteins. NS1 regulates the production of infectious particles [22].
Immunodominant protein E of KFDV, like other flavivirus E proteins, aids in receptor binding and entrance into host cells via the fusion of the viral and cellular membranes [23]. There is evidence suggesting that within the structural proteins of KFDV and other flaviviruses, the E protein plays a crucial role in determining the virus's virulence or tissue tropism [24]. When a virus invades a host, it attaches to the cell’s surface with the help of the E-protein, and the neutralizing antibodies produced in the second week recognize epitopes mostly found in the E-glycoprotein of flaviviruses [25]. Furthermore, computational studies have illustrated that the E protein exhibits significant immunodominance, heightened antigenicity, and a reduced propensity for eliciting allergic responses when compared to other proteins associated with KFD, thereby rendering it an optimal candidate for the advancement of innovative vaccines [26] and methodologies [27].
2. MATERIALS AND METHODS
2.1. Selection of the KFD Strain to Retrieve the Protein Sequence
To identify a suitable strain for this study, the KFD Virus species isolated and gene-sequenced between 1957 and 2020 were obtained from NCBI and analyzed. The data revealed that the strains were isolated and the genes were sequenced from humans, natural hosts, and reservoirs of KFD infection, as well as the vector (tick - Haemaphysalis spp.) [28]. Table 1 presents the details of the KFDV sequences isolated from humans and selected for this study.
| S.No. | Accession Number | Strain | Place of Isolation | Year of Isolation |
|---|---|---|---|---|
| 1 | KY779856.1 | 62957 | Hillemarur, Gadag, Karnataka | 1962 |
| 2 | KY779858.1 | 67965 | Sagar, Shimoga, Karnataka | 1967 |
| 3 | KY779854.1 | NIV12839 | Thirthahalli, Shimoga, Karnataka | 2012 |
| 4 | KY779863.1 | NIV121865 | ||
| 5 | KY779855.1 | NIV12869 | ||
| 6 | KY779859.1 | NIV121863 | ||
| 7 | KP315947.1 | NIV146034 | Malappuram, Kerala | 2014 |
| 8 | ASF57827.1 | MCL-16-H-1297 | Goa | 2016 |
| 9 | MF186846.1 | MCL-16-H-8 | ||
| 10 | MF186845.1 | MCL-16-H-29 |
2.2. Selection of a Suitable Antigenic Protein
Antigenicity refers to the ability of a foreign entity or antigen to be recognized by specific antibodies produced in response to an immune-mediated reaction. The VaxiJen 2.0 server determined the KFD viral protein antigenicity at a threshold of 0.4 [29]. For epitope prediction, the FASTA sequence of the protein with the highest antigenicity score will be selected and saved.
2.3. Cytotoxic and Helper T Lymphocytes Epitopes’ Prediction
To initiate an immune response, antibodies / T-cell receptors bind to the epitope. Adaptive immune responses are elicited by the T-lymphocytes. In eliciting cytotoxic T-cell (CTL) responses, antigens are processed intracellularly, and linear epitopes are primarily targeted. Human Leukocyte Antigen (HLA) plays a significant role, and these antigens are responsible for identifying cells as they enter the body. To develop a vaccine, its epitopes must bind to more than one MHC allele and cover nearly all major populations worldwide. The Allele Frequency Database was used to choose MHC Class 1 HLA-A*24:02 and MHC Class 2 HLA-DRB1 * 15:01 due to the recurring incidence of HLA predominance in the Indian population [30, 31]. The default parameters of epitope prediction programs, such as the IEDB MHC-I tool and NetMHCpan4.1, were used to predict CTL epitopes of nine mer each, whereas the IEDB MHC-II tool was utilized to predict HTL epitopes. 15-mer epitopes with a percentile rank less than 10 were anticipated [32]. The epitopes with the minimum percentile rank exhibit a high level of attraction to MHC-II.
2.4. Linear B-cell Epitope Prediction
B-cell epitopes are necessary for eliciting an antibody-mediated natural defense, which in turn kindles B cells to produce appropriate antibodies. It is predicted using tools such as ABCpred, BepiPred, and Ellipro from the IEDB Analysis Resource [33-35]. A higher peptide score indicates a greater likelihood of becoming an epitope. [36]. Residues that achieve scores above the default threshold of 0.35 are considered as an epitope. [35].
2.5. Antigenicity Prediction of Epitopes
The antigenic nature is determined using VaxiJen2.0 at a cutoff value of 0.4 [29]. This tool was employed to determine the antigenic nature of the predicted 25 CTL&HTL and 66 B-cell epitopes.
2.6. Allergenicity Prediction of Protein Sequences
Prior to developing a vaccination candidate, allergens must be identified. If a protein and recognized allergens share more than 35% of identical sequences over an 80 amino acid window, it is considered to be a possible cause for allergy [37]. Using AllerTop v.2.0, the anticipated epitopes' allergic nature was identified [38]. The chosen epitopes were subjected to analysis using Allertop, and the epitopes that were found to be non-allergenic were further used for the vaccine construct and studies. The same results were cross-checked using AlgPred [39].
2.7. Toxicity Evaluation of Predicted T-Cell Epitopes
The toxicity test is to determine whether a substance has detrimental impacts on human health, animal health, or the environment in general. The toxic nature of the selected epitopes was predicted using the server ToxinPred [40]. The tool generates all the possible mutants of a given peptide and also identifies the toxic regions in proteins [40].
2.8. Immunogenicity Prediction of Predicted T-Cell Epitopes
Immune cells with the capacity to develop pathogen-specific memory, which confer immunological defense, are T and B cells, which facilitate adaptive immunity. For B and T-cells to become memory cells, they must recognize specific targets (antigens) on pathogens through specialized receptors. Henceforth, to design effective vaccines and gain a deeper understanding of the immune system, it is crucial to predict immunogenic CTL and HTL epitopes precisely. Immunogenicity predicting tools available from IEDB were utilized to analyze the immunogenicity of the chosen epitopes. A higher IEDB Class-I MHC prediction score indicates an increased likelihood of eliciting an immunological response. To predict the immunogenicity score of MHC-II/CD4 epitopes, the default parameters are set with a maximum combined score threshold value of 90 [41].
2.9. Prediction of IFN Gamma-Inducing Epitopes
The IFN epitope was used to further investigate the screened 10 HTL-epitopes for their capacity to elicit an IFN-γ immune response [42]. The IFN epitope enables users to determine the peptides or antigens that induce MHC class II binding when exposed to IFN-γ. For the in silico vaccine candidate development, the epitopes that showed positive IFN response outcomes were ultimately selected.
2.10. Conservation Analysis of Predicted T-Cell Epitopes
Measures of identity and conservancy are used to define and characterize epitope or protein variability. It is important to perform a conservancy analysis when estimating epitopes, as it determines whether the epitope is cross-reactive among different isolates of a virus or with different microorganisms that have varying degrees of pathogenicity. We computed epitope linear conservation across antigens using the Epitope Analysis Tools of IEDB, which compute the conservation degree of an epitope within a range of protein sequences at a particular identity level. The tool was employed to shortlist epitopes with a criterion of 100% sequence identity or higher [45].
2.11. Conceptualization of multi-epitope PKFDVac-I (PIIC Vac Candidate-I)
The MEPV sequence was meticulously formulated utilizing innocuous, non-allergenic, and significantly antigenic Cytotoxic T lymphocytes, B-cell epitopes, and Helper T lymphocyte epitopes. These epitopes were interconnected via GPGPG, AAY, and KK linkers to guarantee that the vaccine construct functions as a distinct immunogen and invokes greater concentrations of antibody production than a solitary immunogen [55]. By exploiting AAY as a proteosome cleavage site, a protein’s stability, immunogenicity, and epitope presentation can be altered [56].
KK is used to maintain the immunogenicity of the vaccine constructs [57]. Figure 1 provides a schematic representation of the complete process involved in predicting epitopes and developing PKFDVac-I.
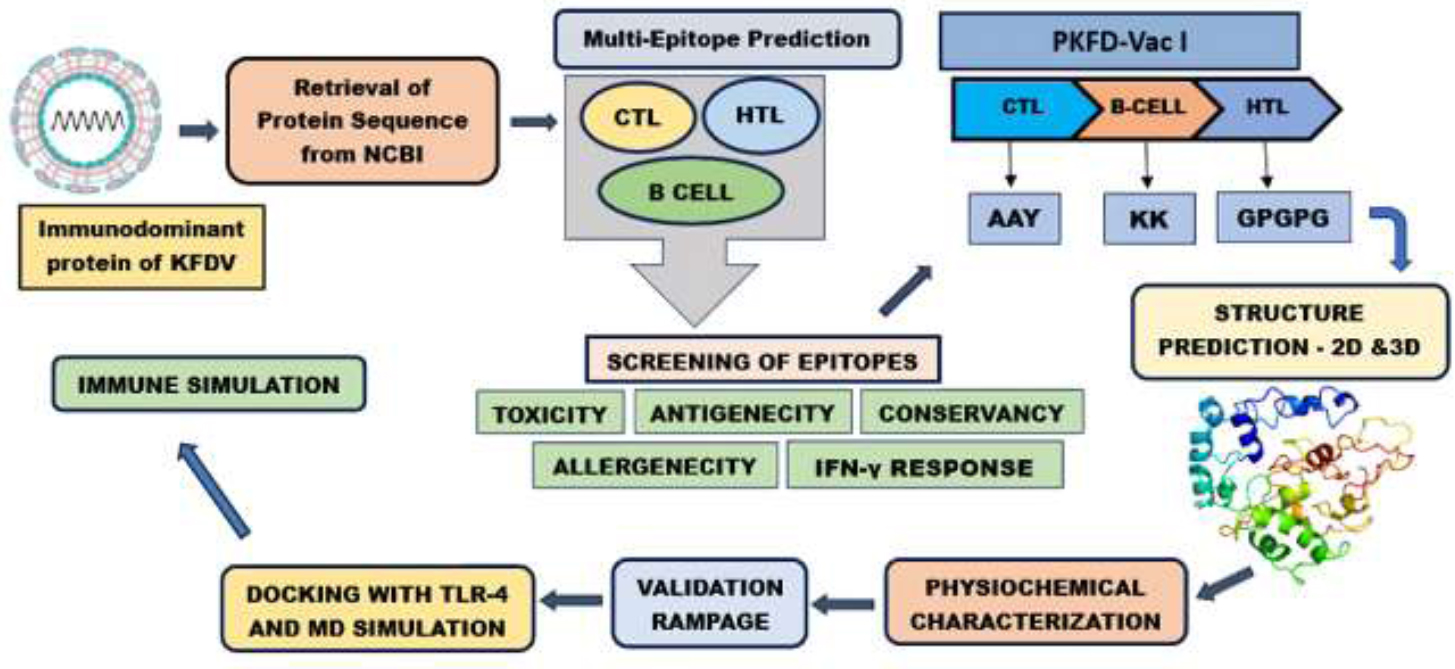
Comprehensive workflow for epitope prediction and development of PKFDVac-I.
2.12. Physicochemical Properties of PKFDVac-I
Vaccine quality attributes of PKFDVac-I were analyzed along with other physicochemical characteristics. The antigenicity was determined using VaxiJen v2.0 at a threshold value of 0.4. The allergenic property was determined using AllerTopv. 20 [38]. Employing the Expasy Protparam tool, the physicochemical attributes were forecasted [46]. The solubility of the MEPV was evaluated utilizing the Protein-Sol server [47].
2.13. 2D and 3D Structure Prediction
The 2D structure of PKFDVac-I was determined using PSIPRED and SOPMA servers. The online tool Psipred 4.0 accurately predicts transmembrane helices, folds, domain recognition, and topology [58]. SOPMA server uses information from a multiple sequence alignment (MSA) of a protein belonging to the same family to predict the 2D structure [49].
The 3D structure of PKFDVac-I was modeled using Alphafold Colab. It is the first computational method that routinely predicts protein structures with atomic exactness, even when there is no known structure identical to it [50].
2.14. Refinement of the 3D Structure and Validation
Validation of the 3D structure is a crucial phase, as it identifies potential errors in models that were predicted. A software PROCHECK with a visual database called PDBsum [51] provides a concise overview of the information contained in each 3D structure submitted to the PDB. The PROCHECK programs are helpful for evaluating the quality of protein structures, both those that have already been solved and those that are being modeled after existing structures.
The model, which scored better in the Ramachandran plot statistical analysis, will be refined and energy minimized. The minimization of the energy of the 3D model was done using Chiron: The rapid protein energy minimization server [52] and upgraded using the Galaxy Refine web server.
2.15. Molecular-level Docking of the PKFDVac-I Vaccine Candidate Construct with the TLR-4 Immune Receptor
The concept of Molecular Docking is based on the understanding that an effective immune response depends on the interaction between an antigen and a specific immune receptor. To examine the interactions between vaccines and receptors, researchers utilized the Maestro program from the Schrödinger suite to conduct a molecular docking study [59]. The tertiary structure of PKFDVac-I was used as a ligand, and the receptor TLR4 (PDB: 4G8A) was downloaded from PDB. The crystallographic structure of the human TLR 4 complex comprises 10 chains designated A through H (A-H). Chains A and B represent the human TLR 4 receptor, chains C and D correspond to lymphocyte antigen 96, and chains E-H are linked to various types of glucopyranose [60]. The vaccine model and receptor were prepared using the Schrödinger Protein Preparation Wizard with default settings. During the process, the hydrogen atoms were added, crystals were removed, and fractional charges were allocated using the OPLS-2005 force field. The binding site of Chain C (MD2) was found to be an active binding site. A grid box measuring (96 x 96 x 96 Å) was selected to surround the Chain-C area, with the grid center located at the center of this previously described binding site. The docking was performed with a Protein-Protein docking module. The top-docked poses were selected as the lowest Glide score. The PDBsum online server was used for analyzing the binding residues and interaction surfaces of the TLR4 and vaccine complexes.
2.16. Molecular Dynamics Simulation
To further examine the interaction between vaccine-receptor complexes, a Molecular Dynamics (MD) simulation was conducted. The simulation was executed using Schrödinger's Desmond module, version 2.3. The System Builder application within Desmond was utilized to prepare the systems for subsequent calculations. An orthorhombic periodic boundary box with a 10 Å buffer distance on all sides of the reservoir was designated to ensure a specific volume. Following the solvation of the protein-vaccine complex within this system, energy minimization and relaxation were carried out using the default protocol available in the Desmond module, employing the OPLS 2005 force field parameters [61]. The simulation was run for 100ns at 300K in an NPT ensemble, with total energy (kcal/mol) recorded every 100 ps. Upon completion of the MD simulation, the stability of the complex was evaluated using Desmond's Simulation Event Analysis tool. This tool analyzed the trajectory file produced by the MD simulation to determine hydrogen bond interactions, RMSF, and RMSD.
2.17. Immune Simulation
To assess the immunological retort of the engineered peptide, immune simulation using the C-Imm sim server was carried out [62]. In pre-clinical testing of the prototype vaccine, shorter intervals between doses are used to quickly elevate antibody titers above protective levels. Clinically, the recommended minimum period between two vaccine doses is one month, or approximately four weeks [63]. For our simulation, we used time step parameters of 1, 84, and 168, corresponding to 8-hour intervals over 1000 simulation steps [64, 65]. The immune response simulation followed the same protocol and default parameters as reported in previous studies. The capability of the PKFDVac-I multiepitope peptide vaccine to stimulate immune system cells, including dendritic cells, immunoglobulins, B-cell lymphocytes, HTL, CTL, and natural killer cells, was studied.
3. RESULTS
3.1. Identification, Analysis of KFD Viral Strain and Retrieval of Immunodominant Protein
The chosen Indian strains' accession codes and the gene sequence encoding their envelope protein were obtained from NCBI and saved in FASTA format. The glycoprotein E of KFDV consisted of 496 amino acids. To date, all tick-borne Flaviviruses analyzed have three potential N-glycosylation sites in the E protein [66]. It helps create a bridge between the host and viral cellular membranes, allowing the virus to enter the host cell. In addition, it induces host immunity by producing neutralizing and protective antibodies [25]. Moreover, structural components of the E protein aid in viral attachment, fusion, hemagglutination, host range, penetration, and cell tropism, as well as viral virulence and attenuation during spontaneous infection or vaccination [67]. E protein plays a significant role in determining virus virulence or attenuation in studies with neutralization-resistant escape mutants and attenuated strains of different Flaviviruses [68] and vaccine development [23, 26].
3.2. Antigenic Evaluation of the Immunodominant Protein of KFDV
The antigenicity score was analyzed, and a comparative study (Table 3) found that all the strains gave an antigenicity score of above 0.45, and the strain with the highest antigenic value was selected for further study. Out of the protein sequence of the ten isolates, strain 62957 and 67965 isolated from humans showed the highest antigenicity score of 0.6649, but to have a species isolated and sequenced recently, MCL-16-H-1297 (0.6634) isolated in the year 2016 was selected over the ones isolated from 1962 and 1967, respectively.
| S.No. | Accession Number | Strain | Antigenicity Score | Antigenicity |
|---|---|---|---|---|
| 1 | ARJ34245.1 | 62957 | 0. 6649 | Probable Ag |
| 2 | KY779858.1 | 67965 | 0. 6649 | |
| 3 | ASF57827.1 | MCL-16-H-1297 | 0. 6634 | |
| 4 | ARJ34248.1 | NIV121863 | 0. 6634 | |
| 5 | MF186846.1 | MCL-16-H-8 | 0. 661 | |
| 6 | MF186845.1 | MCL-16-H-29 | 0. 661 | |
| 7 | ARJ34243.1 | NIV12839 | 0. 6587 | |
| 8 | KY779863.1 | NIV121865 | 0. 6587 | |
| 9 | KY779855.1 | NIV12869 | 0. 6587 | |
| 10 | KP315947.1 | NIV146034 | 0. 6338 |
3.3. Cytotoxic and Helper T Lymphocytes Epitopes’ Prediction
Using in silico tools, the epitopes were predicted and selected based on the percentage rank. A higher score denotes a higher likelihood of triggering an immunological response, and the top 25 CTL epitopes were carefully chosen for further study (Table 4). The lower adjusted rank of the epitopes in IEDB indicates higher binding of the epitopes. Based on the MHC-II (HTL) binding percentile rank, the top 25 epitopes were selected (Table 5).
| S.No. | Start | End | Peptide | Score | Rank | Toxicity | Allergenicity | Antigenicity Score | Antigenicity | Immunogenicity Score |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 131 | 139 | VYDVNKITY | 0.549941 | 0.17 | Non Toxin | AL | 0.2774 | NAG | -0.02556 |
| 2 | 391 | 399 | QWFQKGSTI | 0.408537 | 0.25 | NAL | 0.7212 | PA | -0.3736 | |
| 3 | 384 | 392 | YVGELSHQW | 0.307264 | 0.33 | AL | 0.9755 | -0.09516 | ||
| 4 | 440 | 448 | AFGAAFNTI | 0.276549 | 0.37 | NAL | 0.0389 | NAG | 0.21589 | |
| 5 | 129 | 137 | GYVYDVNKI | 0.245328 | 0.4 | -0.226 | -0.06132 | |||
| 6 | 236 | 244 | NHADRLVEF | 0.208005 | 0.47 | AL | 0.334 | 0.16832 | ||
| 7 | 212 | 220 | AWQVHRDWF | 0.141714 | 0.68 | -0.27 | 0.2323 | |||
| 8 | 219 | 227 | WFEDLSLPW | 0.131912 | 0.71 | NAL | 0.8552 | PA | -0.1275 | |
| 9 | 100 | 108 | GWGNHCGLF | 0.084028 | 0.94 | -0.198 | NAG | 0.00736 | ||
| 10 | 280 | 288 | KYHLQSGHV | 0.063619 | 1.2 | AL | 1.2992 | PA | -0.22169 | |
| 11 | 458 | 466 | ILLGVALAW | 0.058785 | 1.2 | NAL | 1.2625 | 0.12103 | ||
| 12 | 415 | 423 | VVGEHAWDF | 0.055733 | 1.3 | 1.0979 | 0.37998 | |||
| 13 | 207 | 215 | EHLPKAWQV | 0.038136 | 1.5 | AL | -0.068 | NAG | -0.06867 | |
| 14 | 472 | 480 | RNPTLSVGF | 0.037608 | 1.6 | NAL | 1.2659 | PA | -0.07643 | |
| 15 | 157 | 165 | HSNRKTASF | 0.035503 | 1.6 | 0.7897 | -0.18712 | |||
| 16 | 433 | 441 | VGKALHTAF | 0.026538 | 1.9 | 0.5436 | 0.04464 | |||
| 17 | 298 | 306 | KMKGMTYTV | 0.025876 | 1.9 | AL | 0.2284 | NAG | -0.1508 | |
| 18 | 437 | 445 | LHTAFGAAF | 0.018751 | 2.2 | NAL | 0.5545 | PA | 0.25375 | |
| 19 | 178 | 186 | DYGDISLTC | 0.01777 | 2.3 | AL | 2.7368 | 0.02051 | ||
| 20 | 304 | 312 | YTVCEGSKF | 0.014965 | 2.5 | -0.339 | NAG | -0.177 | ||
| 21 | 227 | 235 | WRHEGAQEW | 0.014567 | 2.5 | -0.042 | 0.14182 | |||
| 22 | 446 | 454 | NTIFGGVGF | 0.013139 | 2.6 | NAL | 1.171 | PA | 0.28054 | |
| 23 | 93 | 101 | RRDQSDRGW | 0.013103 | 2.6 | -0.23 | NAG | -0.1861 | ||
| 24 | 247 | 255 | PHAVKMDIF | 0.011547 | 2.8 | -0.098 | -0.22458 | |||
| 25 | 375 | 383 | QLPPGDNII | 0.010769 | 2.8 | AL | 0.0597 | 0.11142 |
| S.No. | Start | End | Peptide | Percentile Rank | Toxicity | Allergenicity | Antigenicity Score | Antigenicity | Immunogenicity Score |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 449 | 463 | FGGVGFLPRILLGVA | 6.9 | Non Toxin | AL | 0.8824 | PA | 97.5893 |
| 2 | 448 | 462 | IFGGVGFLPRILLGV | 7 | NAL | 0.6317 | 96.8397 | ||
| 3 | 476 | 490 | LSVGFLITGGLVLTM | 7.1 | AL | 1.1053 | 96.4429 | ||
| 4 | 474 | 488 | PTLSVGFLITGGLVL | 7.1 | 0.8094 | 97.9807 | |||
| 5 | 477 | 491 | SVGFLITGGLVLTMT | 7.1 | 1.0443 | 96.6517 | |||
| 6 | 475 | 489 | TLSVGFLITGGLVLT | 7.1 | 1.004 | 97.7092 | |||
| 7 | 478 | 492 | VGFLITGGLVLTMTL | 7.2 | 0.9496 | 96.9447 | |||
| 8 | 446 | 460 | NTIFGGVGFLPRILL | 7.4 | NAL | 0.4995 | 97.0931 | ||
| 9 | 447 | 461 | TIFGGVGFLPRILLG | 7.7 | AL | 0.4624 | 97.4095 | ||
| 10 | 450 | 464 | GGVGFLPRILLGVAL | 7.9 | NAL | 0.7564 | 97.8831 | ||
| 11 | 451 | 465 | GVGFLPRILLGVALA | 11 | 0.7004 | 96.6365 | |||
| 12 | 260 | 274 | QTGILLKSLAGVPVA | 11 | AL | 0.4975 | 77.1681 | ||
| 13 | 452 | 466 | VGFLPRILLGVALAW | 11 | NAL | 0.9199 | 96.9207 | ||
| 14 | 480 | 494 | FLITGGLVLTMTLGV | 13 | 0.8491 | 97.2694 | |||
| 15 | 261 | 275 | TGILLKSLAGVPVAN | 13 | AL | 0.4394 | 76.7846 | ||
| 16 | 259 | 273 | DQTGILLKSLAGVPV | 14 | NAL | 0.4274 | 78.1738 | ||
| 17 | 262 | 276 | GILLKSLAGVPVANI | 15 | 0.4321 | 76.8985 | |||
| 18 | 17 | 31 | GTTRVSLVLELGGCV | 15 | AL | 0.2673 | NAG | 99.189 | |
| 19 | 20 | 34 | RVSLVLELGGCVTLT | 15 | 0.7178 | PA | 98.3934 | ||
| 20 | 19 | 33 | TRVSLVLELGGCVTL | 15 | 0.5554 | 98.2113 | |||
| 21 | 18 | 32 | TTRVSLVLELGGCVT | 15 | 0.4494 | 97.9221 | |||
| 22 | 21 | 35 | VSLVLELGGCVTLTA | 15 | NAL | 0.6342 | 98.6761 | ||
| 23 | 482 | 496 | ITGGLVLTMTLGVGA | 16 | 0.9666 | 98.2232 | |||
| 24 | 481 | 495 | LITGGLVLTMTLGVG | 16 | AL | 0.8506 | 97.9242 | ||
| 25 | 426 | 440 | VGGILSSVGKALHTA | 17 | 0.3051 | NAG | 91.3221 |
3.4. Linear B-cell Epitope Prediction
The 496 amino acids long KFDV E protein sequence, when submitted in the ABCPred server with a threshold setting of 0.51, a total of 52 B-cell epitopes (S.No 1-52) of 16 mer were predicted, 10 epitopes of 16 mer (S.No 53-63) were predicted using the Ellipro tool, and 3 epitopes of different lengths were predicted using the IEDB Bepipred tool (S.No 64-66). Since a peptide with a higher score has a greater chance of being chosen as an epitope, the 66 epitopes (Table 6) were arranged in descending order. In order to develop the vaccine candidate, the epitope with the highest score was selected [36].
| S.No. | Epitope Predicted | Start Position | Score | Toxicity | Allergenicity | Antigenicity Score | Antigenicity |
|---|---|---|---|---|---|---|---|
| 1 | PSMETTGGGFVELQLP | 362 | 0.92 | Non Toxin | NAL | 0.749 | PA |
| 2 | PVRAVAHGEPNVNVAS | 341 | 0.91 | AL | 0.5715 | ||
| 3 | YGDISLTCRVTSGVDP | 179 | 0.91 | 1.632 | |||
| 4 | KGSIVACAKFSCEAKK | 110 | 0.91 | Toxin | NAL | 0.7533 | |
| 5 | TLTAEGKPSVDVWLDD | 32 | 0.89 | Non Toxin | AL | 0.0342 | NAG |
| 6 | TYTVCEGSKFAWKRPP | 303 | 0.89 | Toxin | NAL | -0.182 | |
| 7 | TGDYLAANESHSNRKT | 147 | 0.88 | Non Toxin | 0.3361 | ||
| 8 | ASTVCRRDQSDRGWGN | 88 | 0.87 | 0.3639 | |||
| 9 | PAKTREYCLHAKLANS | 53 | 0.87 | Toxin | AL | 1.1122 | PA |
| 10 | KFAWKRPPTDSGHDTV | 311 | 0.87 | Non Toxin | NAL | 0.0271 | NAG |
| 11 | YVGELSHQWFQKGSTI | 384 | 0.86 | 0.7818 | PA | ||
| 12 | TVVGEHAWDFGSVGGI | 414 | 0.85 | 1.2711 | |||
| 13 | YHLQSGHVTCDVGLEK | 281 | 0.85 | AL | 0.9534 | ||
| 14 | YVVKVEPHTGDYLAAN | 139 | 0.85 | 0.4947 | |||
| 15 | PAMGPATLPEEHQAST | 75 | 0.84 | NAL | 0.4027 | ||
| 16 | GRVLEKTRRGIERLTV | 400 | 0.84 | 0.1359 | NAG | ||
| 17 | HVTCDVGLEKLKMKGM | 287 | 0.84 | 0.9336 | PA | ||
| 18 | SVGGILSSVGKALHTA | 425 | 0.83 | AL | 0.3754 | NAG | |
| 19 | FVSGTQGTTRVSLVLE | 11 | 0.82 | 0.8252 | PA | ||
| 20 | SKPCRIPVRAVAHGEP | 335 | 0.8 | 0.8356 | |||
| 21 | TDSGHDTVVMEVTYTG | 319 | 0.8 | 0.683 | |||
| 22 | KKKATGYVYDVNKITY | 124 | 0.8 | 0.3802 | NAG | ||
| 23 | TGILLKSLAGVPVANI | 261 | 0.79 | 0.4508 | PA | ||
| 24 | VLELDKTAEHLPKAWQ | 199 | 0.79 | 0.2056 | NAG | ||
| 25 | TLSVGFLITGGLVLTM | 475 | 0.78 | 0.9475 | PA | ||
| 26 | SVGKALHTAFGAAFNT | 432 | 0.78 | NAL | 0.415 | ||
| 27 | RRGIERLTVVGEHAWD | 407 | 0.78 | AL | 0.2683 | NAG | |
| 28 | KPSVDVWLDDIHQENP | 38 | 0.78 | NAL | -0.3632 | ||
| 29 | HEGAQEWNHADRLVEF | 229 | 0.78 | 0.5485 | PA | ||
| 30 | RGWGNHCGLFGKGSIV | 99 | 0.77 | AL | 0.397 | NAG | |
| 31 | TSGVDPAQTVVLELDK | 189 | 0.77 | NAL | 0.4616 | PA | |
| 32 | FNTIFGGVGFLPRILL | 445 | 0.76 | 0.4096 | |||
| 33 | AGVPVANIEGSKYHLQ | 269 | 0.76 | 0.2893 | NAG | ||
| 34 | PKAWQVHRDWFEDLSL | 210 | 0.76 | 0.0666 | |||
| 35 | VASLITPNPSMETTGG | 354 | 0.75 | AL | 0.4637 | PA | |
| 36 | DRLVEFGEPHAVKMDI | 239 | 0.75 | 0.4363 | |||
| 37 | QSEKTILTLGDYGDIS | 168 | 0.75 | 0.9777 | |||
| 38 | NSKVAARCPAMGPATL | 67 | 0.72 | 1.1882 | |||
| 39 | VELQLPPGDNIIYVGE | 372 | 0.72 | NAL | 0.6526 | ||
| 40 | CLHAKLANSKVAARCP | 60 | 0.7 | AL | 0.9062 | ||
| 41 | LITGGLVLTMTLGVGA | 481 | 0.7 | NAL | 0.8461 | ||
| 42 | LGVALAWLGLNSRNPT | 460 | 0.68 | 1.6057 | |||
| 43 | LGLNSRNPTLSVGFLI | 467 | 0.67 | 1.5677 | |||
| 44 | WFEDLSLPWRHEGAQE | 219 | 0.67 | 0.704 | |||
| 45 | KTASFTTQSEKTILTL | 161 | 0.67 | 0.7068 | |||
| 46 | DIHQENPAKTREYCLH | 47 | 0.66 | Toxin | AL | 0.7688 | |
| 47 | EPHAVKMDIFNLGDQT | 246 | 0.65 | Non Toxin | NAL | 0.6283 | |
| 48 | THLQNRDFVSGTQGTT | 4 | 0.61 | AL | 1.3512 | ||
| 49 | PGDNIIYVGELSHQWF | 378 | 0.61 | NAL | 0.476 | ||
| 50 | NESHSNRKTASFTTQS | 154 | 0.59 | 0.7077 | |||
| 51 | GTTRVSLVLELGGCVT | 17 | 0.57 | AL | 0.3864 | NAG | |
| 52 | VGFLPRILLGVALAWL | 452 | 0.53 | NAL | 0.8507 | PA | |
| 53 | LPRILLGVALAWLGLNSRNPTLSVGFLITGGLVLT | 455 | 0.869 | 0.9546 | |||
| 54 | HAKLANSKVAARCPAMGPATLPEEHQAST | 62 | 0.795 | >50 residues | 0.6608 | ||
| VCRRDQSDRGWGNHCGLFGKGSIVACAKFSCEAKK | |||||||
| 55 | LVEFGEPHAVKMDIFNL | 241 | 0.72 | Non Toxin | AL | 0.345 | NAG |
| 56 | MEVTYTGSKPCRIPVRAVAHGEPNVNVA | 328 | 0.717 | 0.8816 | PA | ||
| 57 | LEKLKMKGMTYTVCEGSKFAWKRPPT | 294 | 0.696 | 0.0035 | NAG | ||
| 58 | LPPGDNIIYVGELSHQWFQKGSTIGRVL | 376 | 0.682 | NAL | 0.6173 | PA | |
| 59 | PSMETTGGG | 362 | 0.642 | 0.7857 | |||
| 60 | TTQSEKTILTLGDYGD | 166 | 0.589 | AL | 0.8537 | ||
| 61 | SVGGILSSVGKALHTAFGAAFNTIFG | 425 | 0.564 | NAL | 0.3818 | NAG | |
| 62 | HEGAQEW | 229 | 0.562 | AL | -0.0624 | ||
| 63 | TQGTTR | 15 | 0.556 | 0.2289 | |||
| 64 | KVAARCPAMGPATLPEEHQASTVCRRDQSDRGWGN | 69 | -1.04 | Toxin | NAL | 0.6496 | PA |
| 65 | DWFEDLSLPWRHEGAQEWNHAD | 218 | -0.69 | Non Toxin | 0.8595 | ||
| 66 | AWDFGSVG | 420 | 0.14 | 2.5292 |
3.5. Epitopes’ Antigenicity Prediction
The antigenic nature of the predicted epitopes at a threshold value of 0.4 was determined and compared. It was found that out of the 25 epitopes predicted, only 12 CTL & 23 HTL epitopes were found to be antigenic (Tables 4 and 5). Out of the 66 B-cell epitopes predicted, only 46 can be considered as probable antigens (Table 6).
3.6. Epitopes’ Allergenicity Prediction
For the 25 CTL and HTL epitopes, allergenicity predictions were made. According to Tables 4 and 5, respectively, 10 HTL and 14 CTL epitopes were non-allergenic. 36 of the 66 B-cell epitopes turned out to be non-allergic (Table 6).
3.7. Epitopes’ Toxicity Analysis
The selected epitopes were subjected to toxicity prediction at a threshold set at 0.5. The results obtained showed that none among the chosen CTL and HTL epitopes exhibited toxicity (Tables 4 and 5), but 5 B-cell epitopes were toxic in nature (Table 6).
A vaccine candidate must be highly immunogenic, highly antigenic, non-toxic, and non-allergenic in order to be effective. Final epitopes were chosen via a stepwise pipeline giving primary weight to MHC percentile rank (≤2% as strong binders), with additional requirements for antigenicity (VaxiJen ≥0.4), non-toxicity, non-allergenicity, and 100% sequence conservancy across strains. IEDB immunogenicity scores were considered supportive rather than exclusionary, consistent with the principle that efficient HLA presentation is the critical first step for T-cell recognition; consequently, some epitopes with strong binding and favorable safety/antigenic profiles were retained despite modest immunogenicity scores [32, 42, 69]. Only nine CTL epitopes and ten HTL epitopes are compatible with the aforementioned, according to the data in Tables 4 and 5. Only 24 B-cells (Table 7) are shortlisted for the vaccine candidate build and additional research because they fit that description, as Table 6 demonstrates.
| S.No. | Epitope Predicted | Antigenicity Score |
|---|---|---|
| 1 | LGVALAWLGLNSRNPT | 1. 6057 |
| 2 | LGLNSRNPTLSVGFLI | 1. 5677 |
| 3 | TVVGEHAWDFGSVGGI | 1. 2711 |
| 4 | HVTCDVGLEKLKMKGM | 0. 9336 |
| 5 | VGFLPRILLGVALAWL | 0. 8507 |
| 6 | LITGGLVLTMTLGVGA | 0. 8461 |
| 7 | YVGELSHQWFQKGSTI | 0. 7818 |
| 8 | PSMETTGGGFVELQLP | 0. 749 |
| 9 | NESHSNRKTASFTTQS | 0. 7077 |
| 10 | KTASFTTQSEKTILTL | 0. 7068 |
| 11 | WFEDLSLPWRHEGAQE | 0. 704 |
| 12 | VELQLPPGDNIIYVGE | 0. 6526 |
| 13 | EPHAVKMDIFNLGDQT | 0. 6283 |
| 14 | HEGAQEWNHADRLVEF | 0. 5485 |
| 15 | PGDNIIYVGELSHQWF | 0. 476 |
| 16 | TSGVDPAQTVVLELDK | 0. 4616 |
| 17 | SVGKALHTAFGAAFNT | 0. 415 |
| 18 | FNTIFGGVGFLPRILL | 0. 4096 |
| 19 | PAMGPATLPEEHQAST | 0. 4027 |
| 20 | LPRILLGVALAWLGLNSRNPTLSVGFLITGGLVLT | 0. 9546 |
| 21 | PSMETTGGG | 0. 7857 |
| 22 | LPPGDNIIYVGELSHQWFQKGSTIGRVL | 0. 6173 |
| 23 | DWFEDLSLPWRHEGAQEWNHAD | 0. 8595 |
| 24 | AWDFGSVG | 2. 5292 |
3.8. Epitopes’ Immunogenicity Analysis
The high-binding-ability epitopes chosen are subsequently subjected to additional testing to predict their immunogenicity. Positive epitope scores indicate high immunogenicity, suggesting a strong capacity to activate T cells and induce cellular immunity. In Tables 4 and 5, the predicted immunogenicity scores are displayed.
3.9. Prediction of IFN-γ Inducing Epitopes
Out of the 10 shortlisted HTL epitopes, only 3 were found to be positive for IFN-γ (Table 8) and, hence, chosen for the vaccine construct.
| S.No. | Epitope | Method | Result | Score |
|---|---|---|---|---|
| 1 | IFGGVGFLPRILLGV | MERCI | Negative | 6 |
| 2 | NTIFGGVGFLPRILL | 4 | ||
| 3 | GGVGFLPRILLGVAL | 10 | ||
| 4 | VGFLPRILLGVALAW | 11 | ||
| 5 | GVGFLPRILLGVALA | 11 | ||
| 6 | FLITGGLVLTMTLGV | SVM | Positive | 0. 21915474 |
| 7 | DQTGILLKSLAGVPV | 0. 4345997 | ||
| 8 | VSLVLELGGCVTLTA | SVM | Negative | -0.12514912 |
| 9 | GILLKSLAGVPVANI | MERCI | 1 | |
| 10 | ITGGLVLTMTLGVGA | SVM | Positive | 0. 17488738 |
3.10. Conservancy Analysis
Conservancy analysis of the carefully chosen T-cell epitopes predicted from the Immunodominant protein E of the KFD virus was carried out, and it is necessary to align a protein epitope with every protein sequence within a given set of sequences to determine how conserved that epitope is. To achieve effective immunization, epitopes must be conserved among strains all over the world. The predicted result shows 100% conservancy for all the selected epitopes isolated from the strain MCL-16-H-1297 across the other shortlisted strains of the KFD virus used in the study.
3.11. Construction of the Multi-epitope Vaccine Candidate PKFDVac-I Using the Selected Epitopes
5CTL, 3HTL, and 8 B-cell epitopes were finalized from the predicted and screened candidates. These were then linked together to form the vaccine construct, PKFDVac-I, with a total length of 279 amino acids. Figure 2 illustrates the schematic representation of this vaccine construct.
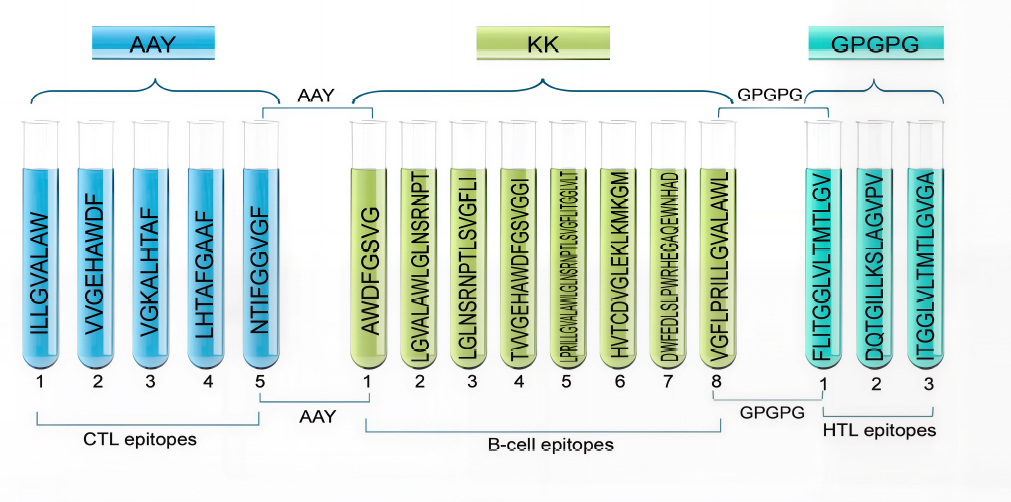
Schematic representation of PKFDVac-I, the multi-epitope peptide vaccine candidate.
3.12. Physicochemical Properties and Solubility Prediction of PKFDVac-I
The protective antigenic score and allergenicity of PKFDVac-I were 0.8882 and -0.81924, respectively, indicating it as a probable non-allergenic antigen based on its amino acid composition [38]. The physicochemical properties, i.e., molecular weight of PKFDVac-I, comprising 279 amino acids, were found to be 29162.32 Daltons. Proteins under 110 kDa are considered better as they can be purified and used for vaccine development more easily [70].
The pI was found to be 9.79. The bulk of the amino acids in the peptide structure have basic structures, evidenced by the comparatively high pI [71]. The atomic composition was elucidated as C1363H2128N350O349S5, comprising 4195 atoms. Assuming that all pairings of Cys residues form cystines, the Ext. coefficient was calculated as 62450 Abs 0.1% (=1 g/l) 2.141. The half-lives were found to be 20 hours for human reticulocytes in laboratory conditions, 30 minutes for yeast in living conditions, and over 10 hours for Escherichia coli in living conditions. The Instability Index (II) is 11.80, indicating that the protein is stable, with an Aliphatic index of 108.39 and a GRAVY score of 0.448. A high aliphatic index indicates good thermal stability, as demonstrated by the new vaccine. The protein appears to be hydrophilic and soluble based on the low GRAVY [71]. Since some amino acids, like alanine, valine, cysteine, and methionine, were found to be under-represented in antibody-antigen recognition sites, while aromatic residues were found to be over-represented, the amino acid composition of PKFDVac-I was extracted to ascertain the percentage of aliphatic and aromatic amino acids present in it [72]. The quality and physicochemical attributes of PKFDVac-I are summarized in Table 9.
| Characteristic Features | Data Report | Inference |
|---|---|---|
| Number of amino acids | 279 amino acids | Suitable for a vaccine candidate[70] |
| Molecular weight | 29162.32 Daltons | |
| Aliphatic index | 108.39 | Increased thermostability. |
| GRAVY | 0.448 | Hydrophobic |
| Antigenicity | 0.8882 | Antigenic |
| Allergenicity | -0.81924 | Non allergen |
| II | 11.80 | Stable |
| Scaled Solubility | 0.554 | Soluble |
The data obtained from Expasy Protparam showed that aliphatic amino acids contribute to 58.9% and aromatic amino acids to 10.1% of the overall amino acid composition. The PsiPred [48] Data obtained while predicting the 2D structure showed that the PKFDVac-I comprised small nonpolar (41.21%), hydrophobic (28.67%), polar (20.07%), and aromatic plus cysteine (10.03%) groups of amino acids, as represented in Fig. (3).
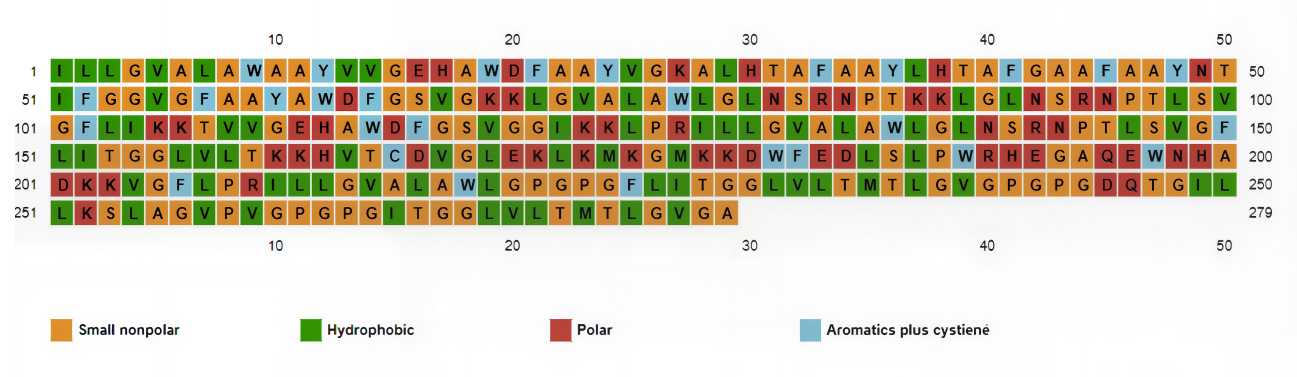
Secondary structure predicted by PSIPRED depicting the amino acid types.
3.13. Secondary Structure Prediction of PKFDVac-I
The 2D structures predicted using PsiPred [48] and SOPMA is shown in Fig. (4A and B). SOPMA revealed 40.50% coiled loops, 25.45% β sheets, and 34% α helices.
Tertiary Structure Prediction of PKFDVac-I
3D Model of PKFDVac-I was predicted with related proteins as the raw input. Five distinct 3D structural models were obtained, and the greater the confidence score, the better the model [50]. The structure with the highest ranking and greatest prediction score was chosen for further refining and validation (Fig. 5A, B) [73].

Predicted 2D structure of PKFDVac-I.
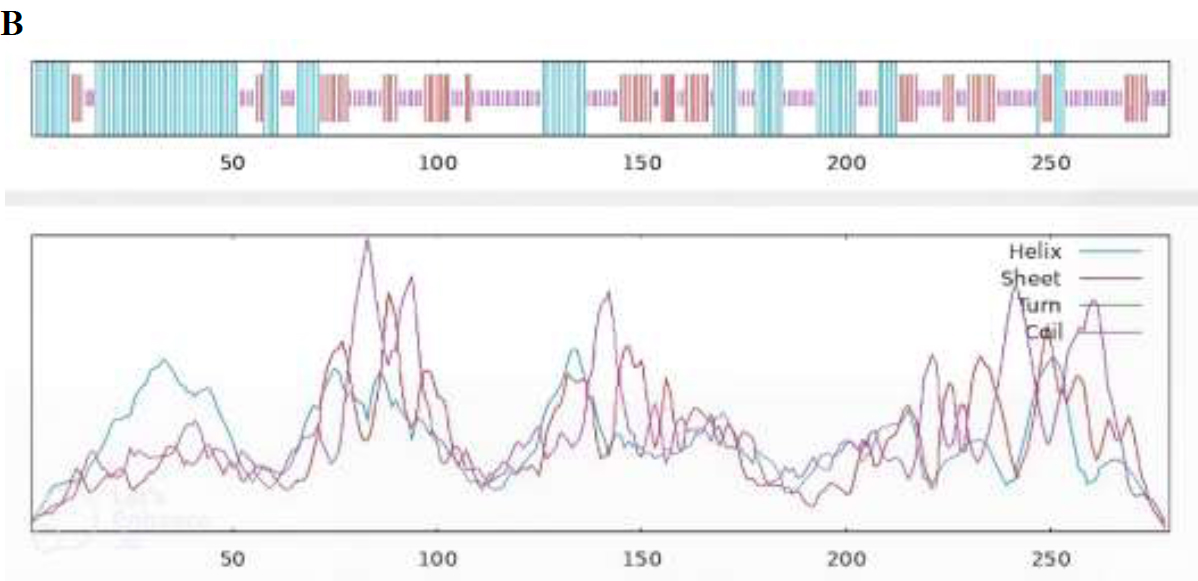
Graphical representation of the 2D structure of PKFDVac-I.
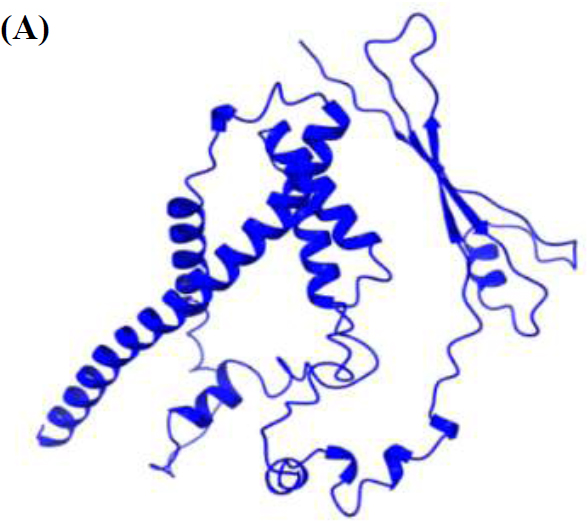
3D structure of PKFDVac-I.
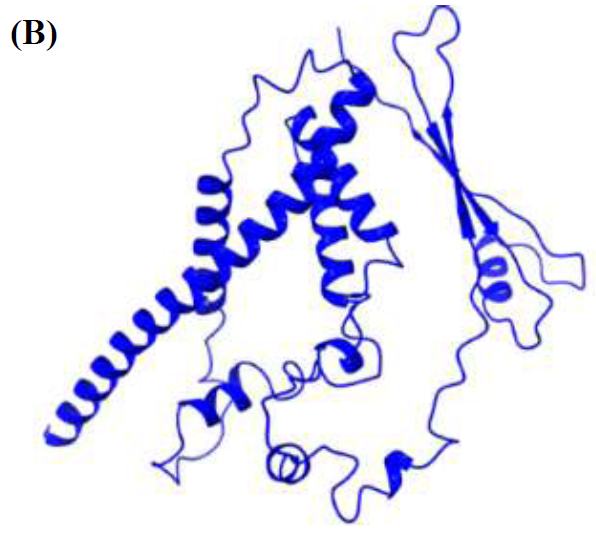
3D structure of PKFDVac-I obtained from Galaxy refine server after the energy minimization and refinement of the structure in Fig. (5A).
3.14. Refinement of the 3D Structure and Validation
When 3D structure refinement was performed, it was found that the VDW (Van der Waals) repulsion energy was minimized to 37.3358 kcal/mol from 173.305 kcal/mol. This was achieved by increasing the number of contacts between atoms to 2274 from 2150 and reducing the number of clashes to 53 from 117. The PDB structure of the refined model, when further subjected to energy minimization using the Galaxy Refine web server, yielded five models with the lowest energy score. The highest Rama favored score, the largest RMSD values, and the lowest energy scores were used to determine which changed structure should be downloaded so that it can be finalized for further studies [57]. According to the results obtained, the amino-acid residues in the Rama-favored regions of the improved model, Fig. (5B-6B) and the original model, Fig. (5A-6A) were 98.6% and 77.3%, respectively. The modified structure's Ramachandran Plot is shown in Fig. (6B), highlighting the improvements made during the refining process. The enhanced structure exhibits more stability, a prerequisite for any potential vaccine candidate, as it is critical to predictability and synthesizability [71].
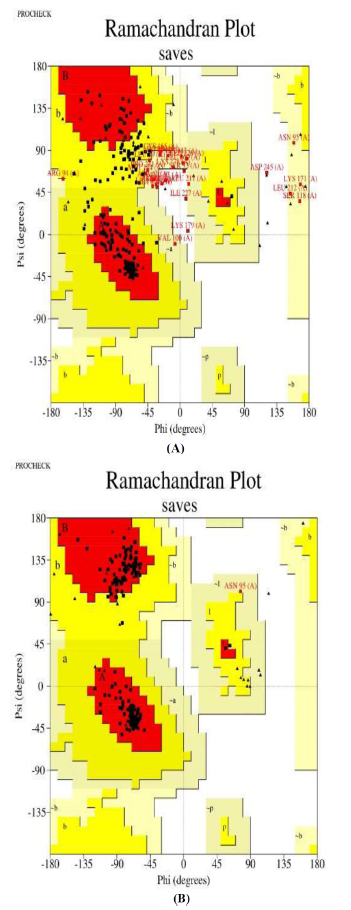
Ramachandra plots of (A) the initial structure and (B) the refined structure of PKFDVac-I. Residues are very weakly concentrated in the favored area, as the original plot illustrates, but a greater concentration is seen in the improved plot.
3.15. Molecular Docking Study
Protein-protein docking was used to determine the stability of the vaccine candidate PKFDVac-I protein receptor complex. The Schrödinger server displayed 30 distinct binding mode poses. On investigating the binding affinity of the vaccine-receptor complex, the top pose with a minimum binding free energy score of -579.716, and Piper energy was -1150.78 was selected. Scores are derived from potential energy changes that occur when proteins and ligands come together. Therefore, a strong binding is indicated by a very negative score, whereas a weak or non-existent binding is indicated by a less negative or even positive score [74]. The complex was then examined through binding interaction residues. The two hydrogen bonds were formed between the Lys91 and Cys95 amino acid residues with the Ala59 residue of the receptor protein, and are represented in Table 10 and Fig. (7). Although only two hydrogen bonds were explicitly identified in the docked PKFDVac-I–TLR-4 complex, numerous non-bonded contacts (hydrophobic packing and van der Waals interactions) and electrostatic complementarity underpin the interface stability, aligning with the highly favorable docking energy. There are no salt bridges formed between two oppositely charged residues, and also no disulfide bonds formed between two cysteine residues. These interactions contribute to the vaccine candidate’s stability by conformational holding of the protein. There was a total of 387 nonbonded contacts that were formed.
Table 10.
| S.No. | Compound | Piper Score | Piper Energy | Interaction Residues | |
|---|---|---|---|---|---|
| Protein | Peptide | ||||
| 1 | PKFDVac-I TLR4 complex | -579.716 | -1150.78 | Lys91, Cys95 | Ala59 |
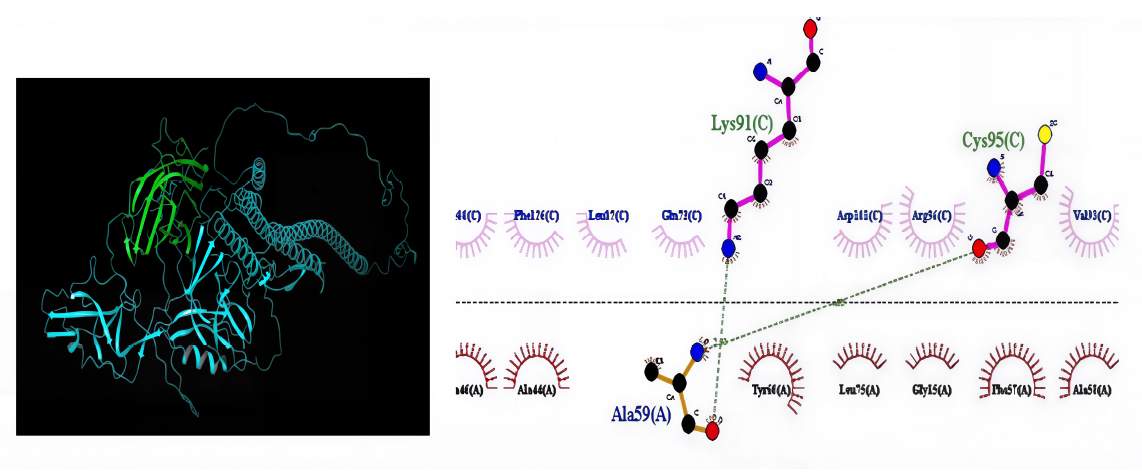
Molecular docking interactions between PKFDVac-I and the TLR4 receptor.
3.16. Molecular Dynamics Simulation
The Desmond v2.3 package of Schrodinger software was used to run an MD simulation for 100 ns. The stability of the vaccine-receptor complex was examined using RMSD and RMSF.
RMSD Calculations
RMSD is the quantitative measure of the similarity between two protein structures [75]. The graphical plot displays the developed vaccine on the right X-axis and the RMSD of the TLR4 protein on the left y-axis.
Initially, the complex was more fluctuated up to 20ns. A slight fluctuation was observed between 20 ns and 40 ns of the simulation time period. Furthermore, after 40 ns, the simulation stabilized, a state that remained consistent until the end of the simulation (100 ns). The overall RMSD plot (Fig. 8) of the complex suggests that it initially fluctuated and then stably bound to the TLR4 binding site, remaining in a constrained position until the end of the 100 ns simulation.
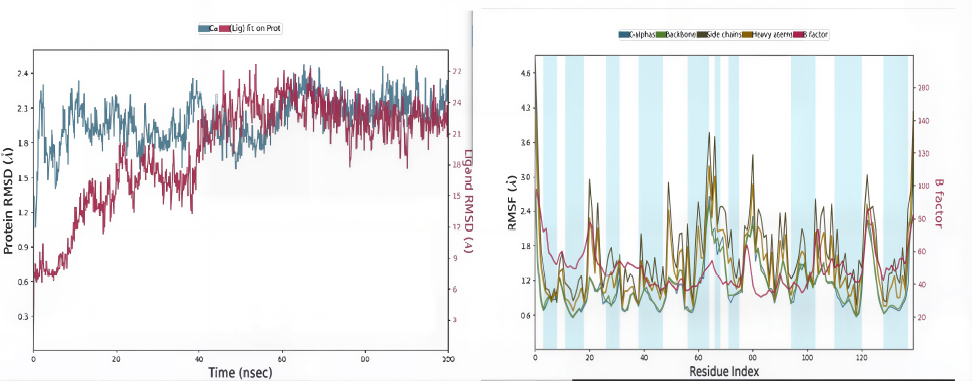
In the RMSD plot (vaccine-receptor complex), blue peaks show protein RMSD and red peaks indicate ligand RMSD. The second image shows the RMSF of the TLR4 protein in the presence of the constructed vaccine, as compared with the B-factor.
Root Mean Square Fluctuation (RMSF) Calculations
A protein's RMSF value is often determined to assess side chain fluctuations caused due to ligand binding [76]. The larger RMSF values indicate flexibility during the simulation period, and the lower RMSF values show good system stability. In Figure 8, red and blue bars indicate the α-helices and β-sheets regions, while the loop area is in white. The plot exhibits higher fluctuations in the C-terminal and N-terminal regions, in contrast to other regions, and minor fluctuations in the RMSF values of the TLR4 protein backbone and side chains in loop regions.
3.17. Immunological Simulation
The immune response to the PKFDVac-I MEPV was evaluated using the C Imm-sim server through in silico simulations. The focus was on the interactions between B-cells, class I and II HLA epitopes, and T-cell receptors with HLA-peptide complexes. Results showed that the vaccine initially triggered an immune response and elicited even stronger responses in subsequent exposures. Higher levels of immunoglobulins and a significant reduction in antigen levels were observed post-vaccination (Fig. 9).
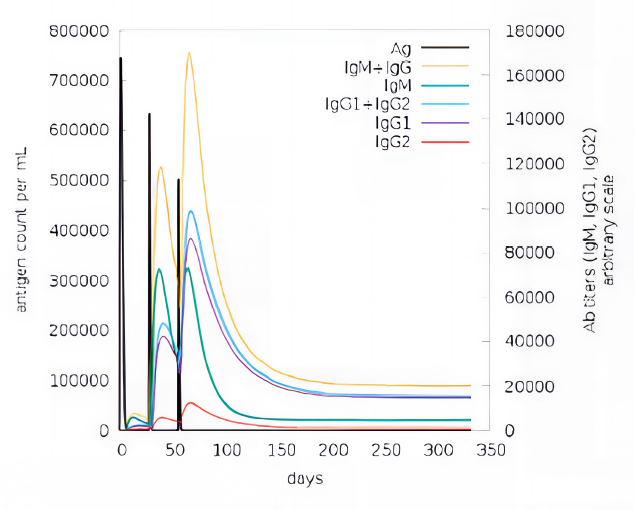
Immune response developed against PKFDVac-I and the levels of immunoglobulins for the antigen concentration using the C-ImmSim server.
The vaccine's effectiveness was evidenced by the increased and sustained B-cell and T-cell populations, with each dose enhancing these levels and promoting the development of memory T-helper and T-cytotoxic cells. These cells remained active throughout the simulation, indicating a prolonged immune response. The T-cytotoxic cell population maintained steady levels of memory cells and increased the number of active cells. Additionally, the increased proliferation of macrophages and dendritic cells suggested efficient antigen processing and presentation to CD4+ and CD8+ cells. The simulation also showed elevated cytokine and interleukin levels, particularly a significant rise in Interferon Gamma, indicating a strong antiviral response generated by the PKFDVac-I vaccine construct.
4. DISCUSSION
KFD, popularly known as Monkey fever, which has been found to have a restricted distribution in Karnataka, India, has recently been expanding its distribution along the stretch of the Western Ghats. Annually, an estimated 400 cases of KFD are documented [19]. According to Kasabi et al. [17] and Holbrook [77], the rate of KFD-infected cases is increasing due to the lack of proper therapeutic methods and the low efficacy of the existing vaccine. The conventional control measures, such as tick control and a CEF-based vaccine, were found to have a limited effect on the cases. Since KFDV is regarded as a highly pathogenic agent and is categorized as BSL4 [78] and KFDV was assigned a Risk Group 4 microbe classification as of 2017, according to the document “Regulations and Guidelines on Biosafety of Recombinant DNA Research and Biocontainment.” [4]. The use of the reverse vaccinology approach has simplified the identification of epitopes that trigger a potent immune response. To develop and formulate the MEPV construct, given the pathogenic characteristics of the virus, the existing genomic sequences of the pathogens were leveraged to predict multi-epitope antigenic peptides, and their physicochemical characteristics were analyzed using immunoinformatic tools listed in Table 2.
Among structural proteins, the KFD virus's Envelope protein (E) is a significant tissue tropism. This protein is vital for the pathogenesis and immune evasion of the virus because it facilitates the virus's entry into the host cell and determines its immunogenetic and phenotypic features [79]. A total of 25 HTLs and CTLs, each, and 66 B-cell epitopes were selected based on their binding score and percentile rank. They were subjected to quality attribute analysis, including antigenicity, allergenicity, toxicity, and immunogenicity.
After comprehensive analysis, 5 CTL, 3 HTL, and 8 B-cell epitopes that met the criteria for a viable vaccine candidate were finalized and linked using suitable linkers to create a stable and effective vaccine construct, potentially leading to a robust IR. The final PKFDVac-I vaccine comprised 279 amino acids with an MW of 29,162.32 Daltons, within the optimal size range for a vaccine. Physicochemical properties revealed an isoelectric point (pI) of 9.79, indicating the pH at which the protein is electrically neutral. The computed instability index (II) is 11.80. suggested protein stability, and the aliphatic index of 108.39 predicted thermal stability [71]. A GRAVY score of 0.448 indicated polarity and hydrophobicity, while a scaled solubility of 0.554 suggested high solubility [46].
Toll-like receptors (TLRs) form the first line of defense against infections and play a pivotal role in adaptive immunity. Specifically, TLRs 1-9, except TLR-5, are involved in responses to viral infections. TLR-4 was selected as an exemplary innate immune receptor, based on its central role in antiviral recognition and evidence from previous research that envelope proteins of flavivirus can engage with TLR-4 to trigger cytokine responses. In addition to the structural role, the E protein functions as a pathogen-associated molecular pattern (PAMP), and its reported interaction with TLR4 highlights the receptor's role as an innate immune sensor, making TLR4 a rational target to study docking interactions and host immune activation in flavivirus infections [79, 80].
In our molecular docking experiments, the proposed vaccine PKFDVac-I candidate demonstrated high binding affinity and low binding energy (Piper energy -1150.78) at the TLR4 receptor binding site. The molecular docking result for TLR4 indicates that the multi-epitope peptide can bind to this central innate immune receptor. This is of biological significance, as the activation of TLR4 is known to induce the production of pro-inflammatory cytokines and type I interferons, which are required for priming adaptive immunity. Such a TLR4-stimulating vaccine candidate would thus enhance antigen presentation, dendritic cell maturation, and subsequent activation of T- and B-cells, eventually culminating in enhanced and long-lasting protective immunity. The 100-ns MD simulation further confirmed a stable complex (consistent RMSD), indicating that the interaction is persistent and biologically relevant.
In normal KFDV-host interactions, viral proteins NS1 and E protein are known to bind to TLR4 in most cases, leading to the overproduction of cytokines and vascular pathology in severe flavivirus infections [81]. In contrast, a well-designed MEPV–TLR4 interaction attempts to leverage the beneficial qualities of TLR4 activation (robust immune priming) without posing the risk of runaway immunopathology. The docking calculations thus provide a rational basis for expecting that the vaccine construct should be capable of replicating natural viral recognition events but in a safer, immunogenic, and controlled manner, supporting its potential efficacy.
The PKFDVac-I, tested using computer simulations, elicited a strong immune response, particularly after repeated doses. The vaccine increased levels of key immune proteins and enhanced the activity of B cells and T cells, which are crucial for fighting infections. These immune cells remained active over an extended period. Additionally, the vaccine boosted the activity of antigen-presenting immune cells, which process and present antigens to other immune cells. Overall, the vaccine generated a potent antiviral response.
Given these promising results, PKFDVac-I warrants further studies through in vitro and in vivo studies to develop a safe and effective vaccine candidate against KFD for human use.
CONCLUSION
KFD is a reemerging zoonotic problem in India, and the resurgence of KFD in India underscores the need for effective preventive strategies that extend beyond conventional vector management. The present study demonstrated the prediction of multiepitope peptide-based immunogens from the virus genome. The analysis has shown the possible application of these immunoinformatic predicted antigens as potential immunogens against KFD. This study proposes that the PKFDVac-I vaccine exhibits high binding affinity and stability, as well as promising immunogenicity in silico, suggesting that it may function as a strong immunogen. This finding highlights the need for further research to evaluate its effectiveness in suitable biological models. The work also emphasizes how immunoinformatically predicted antigens can be chemically synthesized, which makes scaling up for large production simpler. Furthermore, it is worth determining any potential cross-protection provided by these immunogens in vivo, as this could expand the range of uses for them in the fight against related viral illnesses, especially considering the strong genetic resemblance between KFDV and the Alkhurma virus reported in Saudi Arabia.
AUTHORS’ CONTRIBUTIONS
The authors confirm contribution to the paper as follows: S.B.P., S.J.: Conceptualization and design; S.B.P., A.J., S.J.: Methodology; S.B.P., A.J., N.K.P., R.G.P.: Computational work; S.B.P., S.J.: Data analysis; R.S.: Data collection; S.B.P., S.J., P.R.: Writing; S.J.B., P.R.: Reviewing and editing; S.K.C., A.A.P.: Co-ordination of Research; S.J., S.K.C., P.R.: Supervision. All authors read and approved the final manuscript.
LIST OF ABBREVIATIONS
| AHFV | = Alkhurma Hemorrhagic Fever Virus |
| BSL | = Bio Safety Level |
| C Protein | = Capsid Protein |
| CDC | = Centers for Disease Control and Prevention |
| CEF | = Chick Embryo Fibroblast |
| CFR | = Case Fatality Rate |
| CTL | = Cytotoxic T lymphocytes |
| E Protein | = Envelope Protein |
| DMD | = Discrete Molecular Dynamics |
| DNA | = Deoxy Ribo Nucleic Acid |
| FASTA | = Fast Adaptive Shrinkage Threshold Algorithm |
| GRAVY | = Grand average of hydropathicity |
| HTL | = Helper T lymphocytes |
| HLA | = Human Leukocyte Antigen |
| IEDB | = Immune Epitope Database |
| IFN | = Interferon |
| II | = Instability Index |
| IR | = Immune Response |
| KFD | = Kyasanur Forest Disease |
| KFDV | = Kyasanur Forest Disease Virus |
| MD Simulation | = Molecular Dynamics Simulation |
| MEPV | = Multi Epitope Peptide Vaccine |
| MHC | = Major Histocompatibility Complex |
| MW | = Molecular weight |
| NCBI | = National Center for Biotechnology Information |
| NS Protein | = Non-Structural Protein |
| NPT | = Constant number of particles, Pressure, and Temperature |
| PDB | = Protein Data Bank |
| PKFDVac-I | = PIIC Vac Candidate I |
| prM Protein | = Precursor Membrane Protein |
| RMSF | = Root Mean Square Fluctuation |
| RMSD | = Root Mean Square Deviation |
| RNA | = Ribo Nucleic Acid |
| RSSEV | = Russian Spring-Summer Encephalitis Virus |
| TLR | = Toll-like receptor |
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available in the Zenodo at https://doi.org/10.5281/ zenodo.17915145 reference number 2025 (v1).
ACKNOWLEDGEMENTS
The authors would like to thank Ms. Reshma M., Department of Microbiology, Pasteur Institute of India; Dr. Vijayakumar Rajendran, Indian Institute of Science, Bangalore, India; and Mr. Rajesh Nair for their continuous support and guidance.

