All published articles of this journal are available on ScienceDirect.

Functional and Structural Characterization of a Novel Isoamylase from Ostreococcus tauri and Role of the N-Terminal Domain
Authors Info & Affiliations
Abstract
Background:
The debranching starch enzymes, isoamylase 1 and 2 are well-conserved enzymes present in almost all the photosynthetic organisms. These enzymes are involved in the crystallization process of starch and are key components which remove misplaced α-1,6 ramifications on the final molecule.
Aim:
In this work, we performed a functional and structural study of a novel isoamylase from Ostreococcus tauri.
Methods:
We identified conserved amino acid residues possibly involved in catalysis. We also identified a region at the N-terminal end that resembles a Carbohydrate Binding Domain (CBM), which is more related to the family CBM48, but has no spatial conservation of the residues involved in carbohydrate binding.
Results:
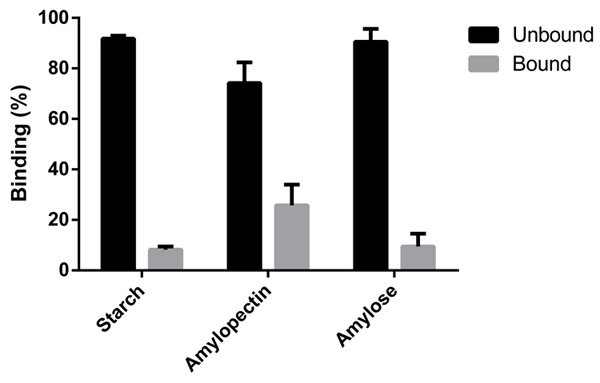
The cloning, expression and biochemical characterization of this N-terminal region confirmed that it binds to polysaccharides, showing greater capacity for binding to amylopectin rather than total starch or amylose.
Conclusion:
This module could be a variant of the CBM48 family or it could be classified within a new CBM family.
1. INTRODUCTION
Carbohydrates are the most abundant molecules on earth; each year photosynthesis fixes approximately 100 billion tons of CO2 and H2O into cellulose, starch, sucrose and other sugars. Starch is one of the most important sources of energy in the human diet. It is composed of two polymers, amylose (essentially an α-1,4-glucan chain) and amylopectin (similar to amylose but branched with α-1,6 glycosidic bonds) whose combination gives rise to a highly insoluble starch granule [1, 2]. Amylopectin represents a 75-90% of total starch content, and approximately 5% of its glucose residues are involved in branch bonds. It was reported that the branching pattern is responsible for amorphous lamellae formation, which is important for the semi-crystalline structure of the granule, and along the linear part of amylopectin molecule, it is responsible for the lamellar organization of starch [3]. In contrast, amylose is present in a smaller amount in the starch granule and is believed to be responsible for granule “filling”, increasing its density [4]. The way in which starch accumulates is controlled by the enzymes involved in its synthesis [5]. The frequency and pattern of amylopectin branching, far from being randomly generated, follow a model that leads to the formation of crystalline lamellae [6]. The ADP-glucose pyrophosphorylase, which synthesizes ADPGlc, is one of the most important enzymes in this process, being the regulatory point of starch synthesis [7-9]. Other enzymes involved in starch synthesis are Starch Synthases (SS) and Branching Enzymes (SBE) [7-9]. Both Granular Bound Starch Synthases (GBSS) and Soluble Starch Synthases (SSS) catalyze the addition of a glucose moiety from ADPGlc to the non-reducing end of the linear α -1,4-glucan chain. While starch synthases generate new α-1,4 bonds, SBE are responsible for the branching of the linear glucan chain, generating α-1,6 glycosidic bonds [10]. On the other hand, Debranching Enzymes (DBEs) participate in the cleavage of the α-1,6 glycosidic linkages generated by SBEs. The latter group of enzymes can be classified into two types: Isoamylases (ISA) and Pullulanases or Limit dextrinases (LDA). While isoamylase can debranch amylopectin, glycogen and phytoglycogen, pullulanase can only act on pullulan and amylopectin [11]. In Arabidopsis thaliana, ISA1 and ISA2 are implicated in amylopectin synthesis, whereas ISA3 and LDA have a major role in starch degradation [1].
Most of the enzymes involved in carbohydrate metabolism comprise a modular organization composed of a catalytic domain and one or more modules named Carbohydrate Binding Domain (CBM) [12]. A CBM is defined as an aminoacid sequence with a particular folding configuration capable to interact and bind to polysaccharides [13]. Although many CBMs are commonly found attached to active enzymes, there are several exceptions where they are part of non-catalytical proteins (e.g. PTST from Arabidopsis thaliana [14]). CBM can be composed of 30 to 200 amino acids and can be as a sole domain or as a part of a tandem. According to their sequence, structure and substrate binding specificity, CBM can be classified into 86 families [15]. They can serve different purposes, and the reason why they are present on the proteins may vary. They can increase the rate of an enzymatic reaction through a strong physical association with the substrate, or they can also act as an organizing subunit, which helps assembling multienzyme complexes [13]. Among CBMs, a particular class is the Starch Binding Domain (SBD). SBDs are found in archaea, bacteria and eukaryotes and are distributed along twelve families of CBM: 20, 21, 25, 26, 34, 41, 45, 48, 53, 58, 68, & 69 (http://www.cazy.org/). These SBD modules have the evolutionary advantage of being able to disrupt the surface of the substrate due to the presence of two different binding sites [16-18].
CAZY is a constantly growing database and hence the number of CBM families is in perpetual growth, as data from new sequencing projects are analyzed and novel CBMs are found. In the last decade, many algal genomes have been sequenced, like those from Micromonas pusilla [19], Ostreococcus lucimarinus [20], Volvox carteri [21], Chlorella variabilis [22], Chlamydomonas reinhardtii [23] and Ostreococcus tauri [24, 25].
Recent effort toward understanding starch metabolism has set O. tauri on the spotlight [26-30]. O. tauri is the tiniest free-living photosynthetic eukaryote known to date and belongs to the ancient Prasinophyceae group [31, 32]. This picoalga has a rather compact genome when compared to other photosynthetic organisms, but it has a large set of genes coding for many proteins involved in starch metabolism. One of the most extreme examples is the case of Starch Synthase III (SSIII) gene: Many plants such as A. thaliana have a single copy of this isoform, while O. tauri has three different copies that codes for SSIII [27, 28, 33-36]. The same is true for Glucan Water Dikinase (GWD) and Phosphoglucan Water Dikinase (PWD); O. tauri has 3 GWD and 2 PWD, while A. thaliana only has 2 GWD and 1 PWD [37]. Another feature of this organism is the presence of a single mitochondrion and chloroplast, that contains a single starch granule [38, 39]. These traits make Ostreococcus an excellent model organism to study starch metabolism and to understand how the fine structure of amylopectin molecule impacts on the final shape of the starch granule through the action of branching and debranching enzymes.
Here, we studied the structural properties of OsttaISA1, an isoamylase coded by Ot14g02550, which is a homolog to ISA1 from Arabidopsis thaliana. We cloned and expressed the N-terminal region of OsttaISA1 to evaluate its function as a CBM, and the interactions with different polysaccharides. In addition, we performed a structural model in order to compare the proposed structure and the location of amino acids important for OsttaISA1 function with other related proteins.
2. MATERIALS AND METHODS
2.1. Cloning, Expression and Purification
The part of the gene Ot14g02550 coding for the putative CBM from O. tauri (named as OsttaCBM) was cloned from genomic O. tauri DNA using primers containing EcoRI and XhoI restriction sites, which are compatible with pRSFDuet-1 expression vector (Novagen, EMD Biosciences Inc, Madison, WI, USA). The procedure was performed using standard molecular biology protocols and the following primers: CBM48I-Fw, ATCGAATTCTCTGGGAGCGACGCGAGTG G (EcoRI site is underlined); CBM48I-Rv, GATCTCGAGT CACTTTGCGTACGGGTCGAGC (XhoI site is underlined).
The plasmid containing the sequence coding for OsttaCBM was transformed into Escherichia coli BL21 (DE3) strain (F- ompT gal dcm lon hsdSB(rB- mB-) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5])). The cells were grown at 37°C to an OD600 = 0.5, and the expression was induced using IPTG to a final concentration of 0,8 mM, and further incubation at 18°C for 14 h. The cells were harvested by centrifugation at 13000 x g for 10 min at 4°C. The pelleted cells were resuspended in a buffer containing 25 mM Tris-HCl (pH 7.5), disrupted by sonication and centrifuged at 13000 x g for 10 min at 4°C. The supernatant was loaded onto a HiTrap chelating HP column (GE Healthcare BioSciences, Little Chalfont, UK) equilibrated with binding buffer (20 mM Tris-HCl pH 7.4, 500 mM NaCl and 25 mM imidazole). The column was washed twice with 15 volumes of binding buffer, and bound proteins were eluted using a linear gradient of elution buffer (25 mM Tris-HCl pH 7.5, 500 mM NaCl and 25-500 mM imidazole) [33]. The presence of the CBM protein in the eluted fractions was confirmed by SDS–PAGE analysis and immunoblotting. The fractions containing the protein of interest with no contaminations were pooled, concentrated to more than 0.5 mg/ml and desalted using Vivaspin 6 3000 MWCO concentrators. Aliquots were stored at -20°C until use after the addition of glycerol to a final concentration of 20% (v/v). Under these final conditions, the protein was stable for at least 3 months.
2.2. Gel Electrophoresis and Immunological Studies
Polyacrylamide gel electrophoresis under denaturing conditions (SDS-PAGE) was done using a Bio-Rad Mini Protean system as described by Laemmli [40] and using 17% (v/v) polyacrylamide/bisacrylamyde gels. Gels were stained after electrophoresis with Coomassie Brilliant Blue R250 (Sigma-Aldrich, St. Louis, MO, USA) or transferred to nitrocellulose membranes (BioRad, Hercules, CA, USA). Electroblotted membranes were incubated with penta-His antibody (anti-His antibody selector kit, Qiagen). The antigen–antibody complex was visualized with alkaline phosphatase-linked a-rabbit IgG, followed by staining with BCIP and NBT as described previously [41].
Native PAGE was performed as previously described by Bollag and Edelstein [42]. Protein samples were prepared in 20 mM Tris-HCl (pH 6.8) containing 10% (v/v) glycerol and 0.01% (w/v) bromophenol blue and then loaded on the gel, which was pre-run for about 2 h at 4˚C (1 h at 100 V, 30 min at 150 V and 1 h at 200 V). Once loaded, the gel was performed at a constant voltage of 200 V at 4˚C using a 10% separating gel (10% (w/v) acrylamide-0.22% (w/v) bisacrylamide, 250 mM Tris-HCl, pH 8.8, 6 x 8 cm x 1mm). Electrophoresis buffer contained 25 mM Tris-HCl and 192 mM glycine (pH 8.8). The gel was then stained using Coomassie Blue.
2.3. Polysaccharide-Binding Assay
Purified OsttaCBM (approximately 15 µg) was mixed with amylose (Fluka-10130), amylopectin (Fluka-10118) or starch (Fluka-85649, St. Louis, MO, USA) in 25 mM Tris-HCl (pH 7.5) at a final polysaccharide concentration of 5% (w/v). The assay was carried out at 20°C under orbital mixing for 20 minutes. The polysaccharides were pelleted by centrifugation at 13000 x g during 10 min and the supernatant was removed and boiled in SDS containing buffer (Tris-HCl 50mM pH 6.8, SDS 2% (w/v), Glycerol 6% (v/v), Bromophenol blue 0.004% (w/v) and β-Mercaptoethanol 1% (v/v)). The pellet was further washed using 25 mM Tris-HCl (pH 7.5) centrifugation, and then resuspended in an equal volume to the supernatant using SDS containing buffer and finally boiled [29, 43-45]. Samples were analyzed on SDS-PAGE and protein levels were determined by densitometric analysis of the protein bands stained with Coomassie Blue R-250, using the software GelPro analyzer (MC, Bethesda, MD, USA).
2.4. Homology Modeling and Sequence Alignment
Sequence similarity search of OsttaISA1 (Genbank code: XP_003083046, 1-851 amino acid residues) whole amino acid sequences was performed with PSI-BLAST [46] with the default parameters (inclusion threshold 0.005) until convergence, using no redundant databases. The sequences with an E value less than 1.10-5 were retrieved and aligned with the program CLUSTALW [47]. Alignment figures were performed using the online program ESPript 3.0 using the default parameters [48]. Phylogenetic tree was built by maximum likelihood using a bootstrap analysis of 500 replicates in MEGA 10.0.2 version [49].
A 3D structural model was obtained using the @TOME V2.3 program, which includes T.I.T.O. (Tool for Incremental Threading Optimization), Scwrl and Modeller [50]. OsttaISA1 structure was modeled using glycogen debranching enzyme Trex from Sulfolobus solfataricus (PDB code 2VR5) [51] and Isoamylase 1 from Chlamydomonas reindhartii (ChlreISA, PDB code 4OKD) [52]. CBM48 was modelled using GlgX from Escherichia coli a template (PDB code 2WSK) [53]. Alignment with the template was based on homology and secondary structure. Models were evaluated using Verify-3D [54] and ProSA-Web [55, 56] online programs. Superposition of the structures was performed using the PyMOL v2.1.0 [57] server v 1.0 [57].
3. RESULTS AND DISCUSSION
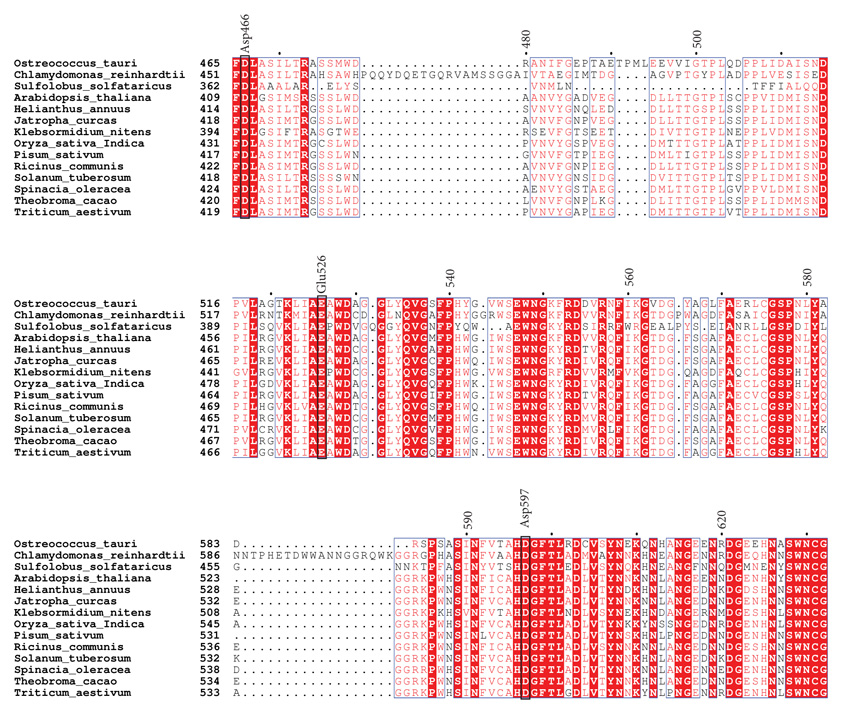
3.1. Sequence Alignment and Phylogenetic Analysis of OsttaISA1
Using PSI-Blast, we conducted similarity searches with OsttaISA1 as a query. Several results were retrieved with E-values lower than 10-5. According to similarity and domain organization, we were able to determine that this protein is strongly related to isoamylases from plants and algae. This bioinformatic prediction allowed us to classify this protein as a family 1 isoamylases-related protein (named as OsttaISA1). The retrieved amino acid sequences were aligned using ClustalW [47], and analyzed using ESPript online server [48]. A partial alignment is shown in Fig. (1) and catalytic amino acids are indicated using the isoamylase from Sulfolobus solfataricus (Trex, PDB code 2VR5) as a template, as the catalytically-important residues have been identified for this organism (numbering refers to OsttaISA1) [51]. It can be observed that Asp363 (Asp466 in OsttaISA1) is conserved not only in OsttaISA1, but also in each analyzed sequence. Other aminoacids that are well conserved are Glu399 (Glu526 in OsttaISA1) and Asp471 (Asp597 on OsttaISA1) Table 1. Numerous studies analyzing the role of these amino acids in the GH13 proteins showed that Asp363 acts as a catalytic nucleophile and would form a covalent intermediate by binding to the glucose moiety at the reducing end of the molecule, while Glu399 is the catalytic acid/base that would protonate the oxygen from the -1 glucose [29, 51, 58]. These aminoacids are involved in the so-called double displacement mechanism, which is proposed for all the reactions involving GH13 enzymes [59]. According to the predictions based on sequence analysis, Asp471 (S. solfataricus numeration) would behave as the transition state stabilizer of the reaction. Additionally, in the characterization of isoamylase I (4OKD - ChlreISA1) from Chlamydomonas reindhartii [52] two different carbohydrate Surface Binding Sites (SBS) have been described. These SBS are non-catalytic regions of the protein that interact with sugar rings through stacking interactions with aromatic residues [60]. Both of these sites, SBS1 and SBS2 are believed to recognize the helical structure of crystalline amylopectin [52]. The residues that provide aromatic rings for the stacking interaction are Trp757 according to 4OKD numbering (Trp740 in OsttaISA1) and Trp767 (Trp750 in OsttaISA1) on site 1, and Trp652 (Trp629 in OsttaISA1), His794 (His778 on OsttaISA1) and Trp856 (Arg840 in OsttaISA1) on site 2. Analyzing complete protein sequence alignment, it can be observed that both Trp from SBS1 are conserved (Trp740 and 750 OsttaISA1 numbering), while SBS2 maintains Trp652 (ChlreISA1) as Trp629 (OsttaISA1) and His794 as His778 (ChlreISA1 and OsttaISA1 numbering, respectively) (Supplementary Fig. 1). The case for the third residue from SBS2, Trp856, is at least unexplored as it is not conserved on OsttaISA1 (Arg840), while the rest of the sequences analyzed in this alignment have an aromatic residue on that position (Table 1). Another feature that is absent on OsttaISA1 but present on ChlreISA1 is the presence of three loops (aa 216 – 226, 466 - 484 and 586 – 604, ChlreISA1 numbering), which are believed to be involved in interface formation on ISA1/ISA2 oligomers on the Chlamydomonas reinhardtii enzyme [52]. Furthermore, it remains to be determined if OsttaISA1 homodimerizes tail-to-tail like ChlreISA1 does, as bioinformatics prediction failed to detect the conservation of the majority of the 48 residues, which are responsible for this interaction. Additionally, a set of amino acid residues that are key to this dimerization are Arg667 and Asp848 (ChlreISA1 numbering). It was described that Arg667 generates a salt bridge between its NH1 with the OD1 of Asp848, however, both residues are absent in OsttaISA1 sequence (Table 1 and Supplementary Fig. 1) [51]. From the amino acid sequence alignment, it can be seen that both residues are not present in the Trex sequence either, suggesting that either these enzymes would not have the ability to form oligomers or that other residues would be involved in this mechanism (Fig. 1).
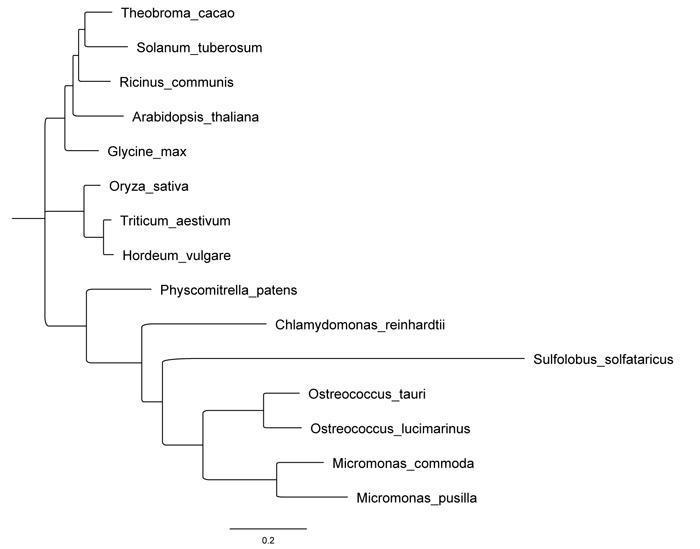
Using the sequences retrieved from BLAST, we performed a maximum likelihood phylogenetic tree (Fig. 2), which demonstrates a clear relatedness of OsttaISA1 to those enzymes from algae. Although the Trex enzyme from Sulfolobus is inside the algae cluster and it seems to evolve from a common ancestor to OsttaISA1, the evolutionary difference between this sequence and OsttaISA1 is clearly evident. Because Trex protein has been crystallized and studied in detail, it is an excellent candidate to obtain OsttaISA1 molecular structure using homology modeling.
3.2. Homology Modeling of OsttaISA1
Using the 3D structure of the previously crystallized 4OKD (ChlreISA1) and 2VR5 (Trex) as a template, we were able to generate a homology model for the isoamylase from Ostreococcus as described in the methods section. The analysis of the structure proposed using 4OKD as a template was evaluated with Verify 3D program and it showed that 98.0% of the residues had a positive score, and 89.6% of them were higher than 0.2 (average score 0.392). Using 2VR5, 99.8% of the residues were greater than zero, while 91.5% were higher than 0.2, averaging on 0.389. The results from Prosa II were also in accordance with high quality models, with its Z-score being -6.45 for 4OKD and -5.68 for 2VR5 [55, 56]. According to these results, we can conclude that the proposed model for OsttaISA1 is of good quality and accuracy to reach confidence in the molecular structure analysis. As described in the previous paragraph, 2VR5 is a glycogen-debranching enzyme with dual activities: α-1,4 transferase and α-1,6 glucosidase, while 4OKD belongs to an isoamylase from the green algae Chlamydomonas reindhartii. Although 4OKD is more related to OsttaISA1 from an evolutionary point of view, Trex has been studied in more depth and allowed us to determine the most important catalytic residues.
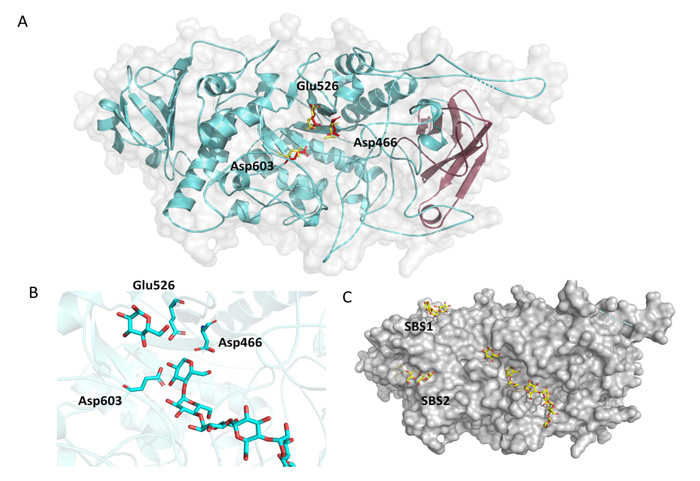
According to protein sequence analysis and classification from InterPro (detailed in Materials and Methods) the region from amino acid 121 to 218 resembles a carbohydrate-binding domain from family 48 (CBM48). Fig. (3A) shows in magenta the position of this putative CBM from OsttaISA1, and it can be clearly observed that it comprises six ß-strands forming the classical CBM ß-sandwich folding [27, 29]. The central part of the protein is composed of the Catalytic Domain (CD), which is also conserved among the α-amylase family, and belongs to the family 13 of glycoside hydrolase from the CAZY database [51, 61]. The CD domain consists of eight parallel ß-strands, which are surrounded by eight α-helices. Furthermore, Fig. (3A) also shows the position of the most important aminoacids for catalysis, being all spatially and rotationally conserved (numbering is referred to OsttaISA1, 2VR5 yellow residues, OsttaISA1 red residues). The zoom-in shown in Fig. (3B) represents the accommodation of the substrate on the cleft and shows the possible interaction with the catalytical triad. Finally, Fig. (3C) shows the surface of the proposed protein folding. It is interesting to observe that both SBS1 and SBS2 have the ability to interact with oligosaccharides as observed for ChlreISA1. The slight curvature of the binding hints the possibility that those SBSs might recognize the helical structure of amylopectin.
3.3. Polysaccharide Binding Assay of the N-Terminal Region of OsttaISA1
Although CBMs can be encountered in non-active enzymes [14], they are normally associated with active enzymes involved in carbohydrate metabolism, driving the substrate to a more intimate and prolonged association with the catalytic domain [13]. It was also demonstrated that this module can act as a macromolecule complex aggregator and can also disrupt the polysaccharide surface [62, 63].
CBM48 can be found both in branching, debranching, and also in non-amylolytic enzymes (e.g. SEX4 protein, SNF1 beta subunit, AMPK beta subunit). They can be on the N- or C-terminal region of a protein and also in the middle of its amino acid sequence, as is the case for SNF1 complex from Saccharomyces cerevisiae [64].
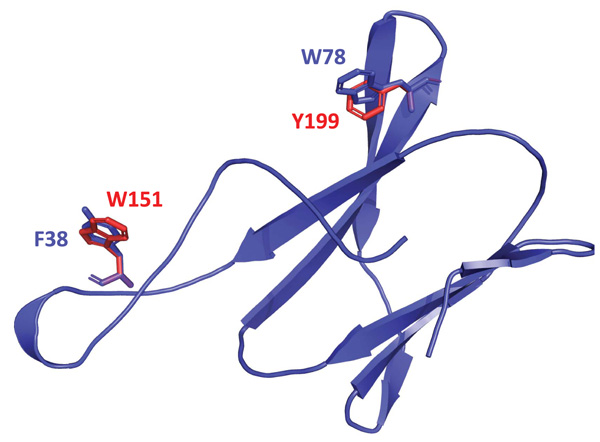
Based on the structure proposed and alignment of the amino acid sequence of the N-terminal end of OsttaISA1 with sequences from other CBMs from family 20, 48 and 53, it was possible to detect amino acid residues that align with those already identified as responsible for polysaccharide binding (Fig. (4) and Supplementary Fig. 1). Fig. (4) shows that W151 and Y199 residues are spatially conserved, but it is interesting to note that the orientation of their aromatic rings differs from those from the amino acids present in the GlgX protein.
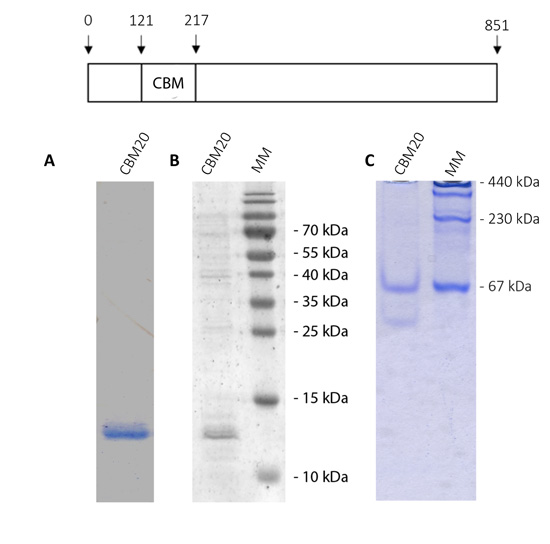
Thus, it is necessary to test if this N-terminal domain would be involved in the binding to polysaccharides. To evaluate this possibility, we cloned the sequence coding for the putative CBM module and expressed it in E. coli cells. Fig. (5 shows the analysis of purified OsttaCBM by SDS-PAGE, western-blotting and native PAGE. A protein band of about 13 kDa corresponding to Ostta CBM can be observed (Fig. 5A and B). As mentioned in section 3.1, we did not find amino acids involved in protein dimerization as has been previously described for ChlreISA1. To analyze whether the OsttaCBM protein has the ability or not to dimerize, we performed a native PAGE. Results show that OsttaCBM has the ability to oligomerize, forming at least a dimer and a 5-subunits protein (Fig. 5C).
These results agree with our bioinformatic analysis, as we predicted two surface binding sites that accommodate polysaccharides with helical structure as is the case of amylopectin. On the other hand, the lower affinity for amylose seems to be related to the linear structure of the molecule. The behavior with starch might respond to a steric hindrance, as seems to be the case of CBM41 from the branching enzyme from Ostreococcus tauri [29].
The purified protein allowed us to further perform polysaccharide binding studies. As Fig. (6) shows, OsttaCBM is able to bind the three substrates analyzed: amylose, amylopectin and starch; however, it can be seen that the greatest binding capacity is for amylopectin.
| Residue | Function | Conserved Function |
|---|---|---|
| Asp363 (Trex) | catalytic nucleophyle | YES (Asp466) |
| Glu399 (Trex) | catalytic acid/base | YES (Glu526) |
| Asp471 (Trex) | transition state stabilizer | YES (Asp597) |
| Trp757 (ChlreISA1) | stacking interactions SBS1 | YES (Trp740) |
| Trp767 (ChlreISA1) | stacking interactions SBS1 | YES (Trp750) |
| Trp652 (ChlreISA1) | stacking interactions SBS2 | YES (Trp629) |
| His794 (ChlreISA1) | stacking interactions SBS2 | YES (His778) |
| Trp856 (ChlreISA1) | stacking interactions SBS2 | NO (Arg840) |
| Arg667 (ChlreISA1) | key for dimerization | NO (Ala648) |
| Asp848 (ChlreISA1) | key for dimerization | NO (Lys832) |
CONCLUSION
OsttaISA1 is classified as a debranching enzyme as its sequence has a very high identity to other debranching enzymes analyzed. According to our alignment and prediction, OsttaISA1 has every amino acid responsible for α-1,6- transferase activity, but lacks a key structural lid region believed to be involved in α-1,4- transferase activity located close to the active site [51]. As previously shown, these lids located close to the active site are key on the latter transferase activity, and in some polypeptides, the lid may be formed through dimerization of the protein [65, 66]. As predicted by our in silico analysis, OsttaISA1 lacks specific residues previously reported to be involved in dimers or higher oligomers formation [51, 52]; however, results show that OsttaCBM is able to form oligomers, at least in vitro. It is clear that further experiments are needed to probe the possible oligomerization capacity of OsttaISA1 and its functional relevance.
The binding assays give new insight into the function of the N-terminal region of the polypeptide. According to the bioinformatic prediction, this domain, which has a greater capacity for amylopectin binding, could be from the CBM family 48 or a variant of this family, because, as mentioned before, although important amino acids are conserved, there was no spatial co-localization for W151 and Y199 (which are responsible for polysaccharide binding) with those from the CBM48 model. These differences in the binding site could be a consequence of structural discrepancies between amylopectin and glycogen.
It is important to note that debranching enzymes are subdivided into two groups according to substrate specificity and protein sequence: ISA and Limit Dextrinase (LDA) [1]. Both Arabidopsis thaliana and Ostreococcus tauri have three ISA proteins: ISA1, ISA2 and ISA3, while there is only one LDA. As has been reported for Arabidopsis, ISA1 (homolog to OsttaISA1) and ISA2 are responsible for the process of debranching activity during the synthesis of starch, while ISA3 and LDA are involved on starch degradation by cleaving α-1,6 bonds [37]. ISA1 and ISA2 might be involved in the so-called “glucan trimming” model, where they correct the misplaced α-1,6 bonds by cleaving the ones that will not allow amylopectin packing into a highly ordered structure [67]. From this point of view, the result obtained is interesting, where amylopectin is the polysaccharide that showed the highest affinity to OsttaCBM. Taken together, these results might explain part of the mechanism that facilitates the cleavage of certain bonds but not others, generating the crystalline starch granule structure this way. In addition, the characterization of new enzymes capable of conferring differential properties to starch is of high importance to eventually design new strategies to obtain novel starches for different uses in different industries.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
This work was supported by grants from CONICET (PIP 0476), ASaCTeI (IO-2018-00098) and ANPCyT (PICT 2018-01440). NH is a research fellow from ANPCyT. JB, DFGC and MVB are research members from CONICET.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We are grateful to María Belén Velázquez for excellent technical assistance.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.

