All published articles of this journal are available on ScienceDirect.

De Novo Assembly and Transcriptome Profiling of Ethiopian Lowland Bamboo Oxytenanthera Abyssinica (A. Rich) Munro Under Drought and Salt Stresses
Authors Info & Affiliations
Abstract
Background:
Bamboos are perennial grasses classified under family Poaceae and subfamily Bambusoideae and are among the fastest growing plants on earth. Despite ecological and economic significances, Ethiopian lowland bamboo (O. abyssinica) lacks global gene expression under abiotic stress.
Methods:
Plastic pot germinated seedlings of O. abyssinica were subjected to 200 mM NaCl and 25% PEG-6000 (Polyethylene glycol) to induce salt and drought stress, respectively. Using the Illumina sequencing platform, fifteen cDNA libraries were constructed and sequenced to generate the first drought and salt stress transcriptome profiling of the species so as to elucidate genome-wide transcriptome changes in response to such stresses.
Results:
Following quality control, 754,444,646 clean paired-ends reads were generated, and then de novo assembled into 406,181 unigenes. Functional annotation against the public databases presented annotation of 217,067 (53.4%) unigenes, where NCBI-Nr 203,777, Swissport 115,741, COG 81,632 and KEGG 80,587. Prediction of Transcripts Factors (TFs) have generated 4,332 TFs organized into 64 TF families. Analysis of Differentially Expressed Genes (DEGs) provided 65,471 genes where 569 genes belong to all stresses. Protein families with a higher number of differentially expressed genes include bZIP (49), WRKY (43), MYB (38), AP2/ERF (30), HD-ZIP (25) and MYB related (21).
Conclusion:
In addition to revealing the genome-wide level appraisal of transcriptome resources of the species, this study also uncovered the comprehensive understanding of key stress responsive protein-coding genes, protein families and pathways which could be used as the basis for further studies.
1. INTRODUCTION
Bambusoideaes are composed of 75 genera and about 1, 250 species of bamboos distributed in a range of environments from tropical and warm ecosystems to cold regions [1]. On the basis of morphological habit, Bambusoideaes are classified as woody and herbaceous bamboos [2]. Woody bamboos are known by their lignified culms, specialized culm leaves, bisexual flowers, outer ligules on the foliage leaves, complex vegetative branching and gregarious monocarpy [3]. Herbaceous bamboos are known by short and more frailly lignified shoots, smaller branches, unisexual flowers, cyclic flowering pattern [3]. Only two species of Bamboos, namely Arundinaria Alpina (K.schum) and Oxytenanthera abyssinica (A.Rich) are found in Ethiopia. It is believed that 60% of African Bamboo resources are found in Ethiopia [4].
O. Abyssinica grows at an altitude range of 700 - 1,800 m above sea level in the western part of Ethiopia adjacent to Sudanese Savannah Woodlands. According to FAO and INBAR [5], the major portion of Ethiopia’s bamboo (85%) is the O. abyssinica. One of the important features is, it's a hardy species on poor soils in dry vegetation formation. It is most resistant to drought as it tolerates rainfall down to 700 mm and a high temperature of up to 43oC [6]. O.abyssinica has wider applications, including but not limited to housing, fodder, beehives, pulping and paper, flooring and furniture, charcoal, fiber and textile, plywood for truck carriage and molding board for concrete [7, 8].
O. abyssinica is clump-forming sympodial bamboo with a strong rhizome up to 10 cm in diameter grows in pure stands. The culms/stems are grouped into large dense clumps erect and leaning with a length up to10 m [6]. The base diameter varies from 3-5 cm. During shooting, culms are solid and later on develop into a small central cavity with a thick culm wall. Due to this, O. abyssinica is not easier to split for weaving as A. alpina. The stripped leaves are about 20 cm long and during unfavorable environmental conditions, they have deciduous foliage. There is a morphological difference as some clumps have exceptionally thinner culms and others with much taller and thicker culms among the normal sized ones. Such differences could result from cross-fertilization when flowering. It leads to a variety of seedlings with different growth characteristics of the individual clumps, quite contrary to the monopodial A. alpina [8]. Under conducive natural conditions, it grows at a density of 8000 stems/ha. The above-ground biomass is about 20 tonnes/ha [9].
The basic chromosome number of herbaceous bamboos is 11 (x = 11), while the basic chromosome number of woody bamboos is believed to be 12 (x = 12.) Woody bamboos have different ploidy levels, the hexaploid (2n = 6, x = 72) tropical woody bamboos and the tetraploid (2n = 4x = 48) temperate woody bamboos [10].
In response to serious environmental stresses, a series of plant genes with diverse functions are either repressed or enhanced [11-13]. Proteins coded with stress-induced genes play a significant role during the occurrence of stresses since they are functional and regulatory proteins. Plant Transcription Factors (TFs) are regulatory proteins which control gene expression in reaction to a range of stresses including drought and salinity. Thus, TFs significantly engage in plant growth and development by regulating defense response and gene regulation networks [14]. In plants like rice [15], and Arabidopsis thaliana [16], many genes have been found to be regulated in common or in particular under drought and salt stress. However, abiotic stress-induced transcriptome studies of O. abyssinica have not been undertaken. This study was conducted to unveil the most represented genes, transcript factor families and pathways of the species in response to drought and salt stresses.
2. MATERIALS AND METHODS
2.1. Plant Materials and Treatment
O. abyssinica seeds used in this research were obtained from Ethiopian biodiversity institute collected from Asossa area, West Ethiopia. Seedlings were germinated and grown under plastic pot at 26°C/22°C (day/night) with 75% relative humidity, 16 hr photoperiods and 175 μmol/ (m2. s-1) light intensity. One month grown seedlings with a height range of approximately 35 cm were transferred to water media for one week to create similar condition before treatment. A total of 90 seedlings (30 seedlings per treatment, 10 seedlings per replication) were used for control, salt and drought stress. The seedlings were treated with 200 mM NaCl and 25% PEG-6000 (Polyethylene glycol) to induce salt and drought stress, respectively. The three biological replicates for 24 h treatment were named as W1, W2, W3 for control, N1, N2, N3 for salt and P1, P2, P3 for drought, while the 48 h treatment were W4, W5, W6 for control and N4, N5, N6 for salt. Seedlings treated with 25% PEG-6000 wilted and could not be used for RNA extraction at 48h. After treatment, 15 independent leaves samples (six control, six salt and three droughts) were collected and directly frozen with liquid nitrogen and stored at -80 oC until use.
2.2. RNA Extraction, cDNA Synthesis, and Sequencing
RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Total RNA of each sample was qualified and quantified using NanodropLite spectrophotometer (Thermo Scientific Wilmington, Delaware, USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) with 1% agarose gel. For library construction, one μg total RNA with RIN value above seven was used. Next-generation sequencing library was constructed according to the manu facturer’s protocol (NEBNext Ultra™ RNA Library Prep Kit for Illumina).
NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB) was used to isolate poly(A) mRNA. The mRNA fragmentation was carried out using NEBNext First-Strand Synthesis Reaction Buffer, while priming was conducted using NEBNext Random Primers. First strand cDNA was synthesized using ProtoScript II Reverse Transcriptase and the second-strand cDNA was synthesized using Second Strand Synthesis Enzyme Mix. The purified double-stranded cDNA by AxyPrep Mag PCR Clean-up (Axygen, Union City, USA) was then treated with End Prep Enzyme Mix to repair both ends and add a dA-tailing in one reaction, followed by a T-A ligation to add adaptors to both ends. Size selection of Adaptor-ligated DNA was then performed using AxyPrep Mag PCR Clean-up (Axygen, Union City, USA), and fragments of ~360 bp (with the approximate insert size of 300 bp) were recovered. All samples were then amplified by PCR for 11 cycles using P5 and P7 primers, with both primers carrying sequences which can anneal with flow cell to execute bridge PCR and P7 primer carrying a six-base index letting for multiplexing. The PCR products were purified using AxyPrep Mag PCR Clean-up (Axygen, Union City, USA), confirmed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA), and quantified by Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). Then libraries with different indices were multiplexed and loaded on an Illumina HiSeq instrument according to manufacturer’s instructions (Illumina, San Diego, CA, USA). Sequencing was carried out using a 2x150 bp Paired-End (PE) configuration; image analysis and base calling were conducted by the HiSeq Control Software (HCS) + OLB + GAPipeline-1.6 (Illumina) on the HiSeq instrument.
2.3. Data Filtering and Assembly
Quality assessment of the raw paired-end reads was performed using FastQC v0.11.2 [17]. Pre-processing of raw reads was conducted using Cutadapt v.1.9.1 [18] for adaptor trimming and Sickle v1.33 [19] for quality filtering. Reads obtained after filtering were then used for transcriptome assembly using Trinity v2.2.0 [20]. Trinity works by integrating three different software modules: Inchworm, Chrysalis, and Butterfly execute one after the other to process bulky volumes of RNA-seq reads into full-length transcripts. The duplicated contigs were removed using CD-HIT v4.5.4 [21]. The longest transcripts were taken to be unigenes for functional annotation by identifying nucleotide sequences of all transcripts.
The raw sequencing reads are deposited in the NCBI Sequence Read Archive (SRA) under SRP153816 accession number and the assembled transcripts are also deposited in the Transcriptome Shotgun Assembly (TSA) Database, under GGTK00000000 accession number.
2.4. Functional Annotation and Classification of De Novo Assembled Unigenes
The de novo assembled O. abyssinica unigenes were annotated through homology searches against public protein databases with an (E-value cut-off of 10-5). The unigene sequences were annotated by using Blast2go [22]. The databases used for annotation include NCBI-Nr, COG, Swissprot, KEGG and GO. GO-Term Finder was used for identifying Gene Ontology (GO) terms that annotate a list of enriched genes with a significant P-value < 0.05. The GO and COG functional classification analyses of all unigenes provide valuable information in predicting possible gene functions and in determining the gene function distribution of O. abyssinica unigenes. The KEGG database which deals with genomes, biological pathways, and chemical substances was used to investigate the gene product during plant metabolism and associated gene functions in cellular processes [23].
2.5. Differentially Expressed Genes (DEGs) Analysis
For expression analysis, the unigene sequence file as a reference gene file, RSEM v1.2.6 [24] was employed to guesstimate gene and isoform expression levels from the pair-end clean data.
FPKM (Fragment per Kilobases per Million reads) was calculated and employed to quantify the expression abundance of transcripts in each sample. For differential expression analysis, the DESeq2 v1.6.3 [25] R program, a model based on the negative binomial distribution was used for determining differential expression from digital gene expression data. A DESeq2 analysis was performed using three combinations: (i) control vs. drought, (ii) control vs. salt and (iii) drought vs. salt. To control the false discovery rate, P-value was adjusted by Benjamini and Yekutieli’s approach [26]. Genes with |log2 Fold change| > 1 and adjusted P-value < 0.05 were treated as differentially expressed.
2.6. Validation of RNA-Seq Data by RT-qPCR
To confirm RNA-Seq data by RT-qPCR 40 candidate, unigenes (20 up-regulated and 20 down-regulated) were randomly selected. After removing conserved regions using Expasy (https://www.expasy.org) from the protein sequences, 48 qPCR primers were designed from the full-length cDNA sequences of each unigene using Primer quest tool (supplementary file 1). Total RNA was extracted using TaKaRa MiniVEST Plant RNA Extraction Kit (Takara, Dalian, China). The quality and quantity of total RNA were checked by 1% agarose gel electrophoresis and NanoDrop 2000c Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The RT-qPCR was performed on an ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA) using the SYBR Premix Ex Taq™ kit (Takara, Dalian, China), according to the manufacturer’s instructions. The amplification protocol comprised a 5-minute incubation at 94 °C then a cycle of 94 °C for 30 s, 60 °C for the 30s, 72 °C for 1 min, repeat 35 times, 72 °C for 10 min, and a final 4 °C hold. Relative expression of RT-qPCR was calculated 2-ΔΔCT.NTB- FTCTTGTTTGA CACCGAAGAGGAG and NTB-R AATAGCTGTCCCT GGAGGAGTTT primers from ERF76 gene that was confirmed to be used as references for qRT-PCR from Moso bamboo [27].
3. RESULTS
3.1. Sequencing, De Novo Assembly and Functional Annotation of O. Abyssinica Transcriptome
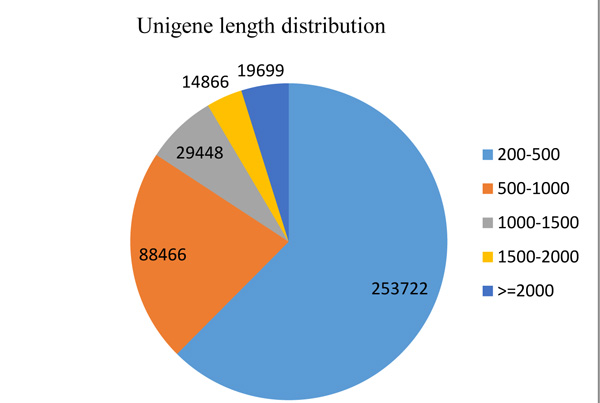
The result of Illumina paired-end sequencing and de novo assembly of O. abyssinica is presented in Table 1 and the length distribution of 406,181 unigenes is illustrated in Fig. (1).
As there was no genomic and transcriptome information published so far on O. abyssinica, NCBI-Nr blast search of the unigenes was conducted against other species. Accordingly, Oryza sativa japonica was identified to be the species with the highest homology (41,400 unigenes, 10.2%), followed by Brachypodium distachyon (25,886) and Setaria italica (21, 881). Other species with considerable matches include Oryza brachyantha (18,081), Zea mays (15,194), Sorghum bicolor (12, 697), Oryza sativa indica (9,504), Aegilops tauschii (8,162), Triticum Urartu (7,393) and Phyllostachys edulis (6, 395).
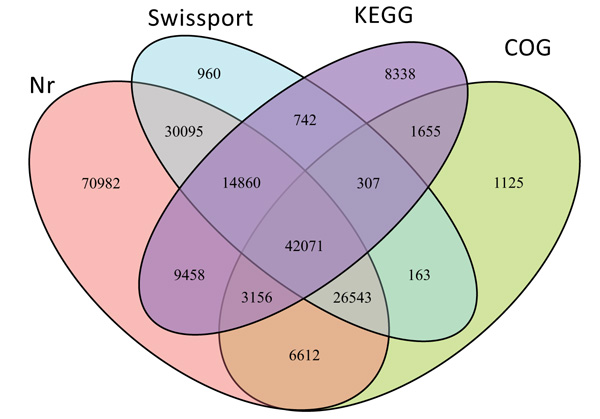
From the total 406,181 identified unigenes 217,067 (53.4%) were successfully annotated using BLASTX searches against the public databases of NCBI-Nr (203,777, 93.8%), Swissprot (115,741, 53.3%), COG (81,632, 37.6%) and KEGG (80,587, 37%). But the remaining 189,114 (46.6%) unigenes did not show any significant functional similarity to any of the database explored. The detailed distribution of annotated unigenes to each and multiple databases is shown in Fig. (2).
3.2. GO and COG Annotation Analysis
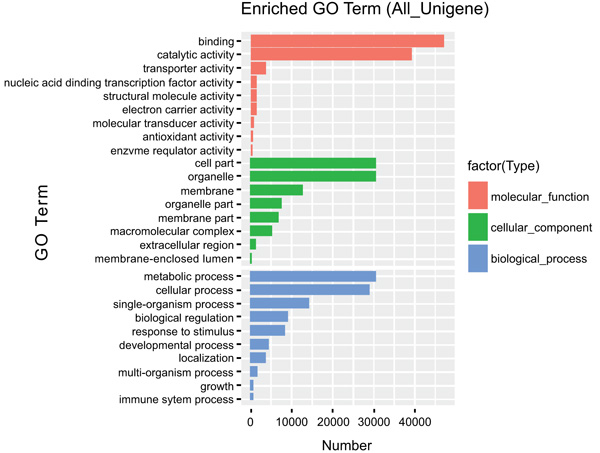
The most over-represented GO terms in response to drought and salt stresses were “binding, 48,000 unigenes”, “catalytic activity, 39,000 unigenes” both under molecular function category. “Cell part, 30,000 unigene”, “organelle 28,000 unigenes” from Cellular component and “Metabolic processes 30,000 unigene”, “cellular process 28,000 unigenes” from the biological process was also represented with a significant amount of unigenes (Fig. 3).
Clusters of Orthologous Groups (COG) database annotation searches of 406,181 unigenes have predicted 81,632 unigenes (20%) that were assigned into 25 functional categories. The most dominantly represented functional subcategories are “Signal transduction mechanisms” with 12,861 (15.7%) unigenes followed by “Posttranslational modification, protein turnover, and chaperones” with 10,191 unigenes (12.5%) which belongs to cellular processing and signaling pathway categories which makes 28% of the total functional annotations. The third most represented is “General function prediction only” with 9993 unigenes (12.2%) followed by “Intracellular trafficking, secretion and vesicular transport” with 4968 unigenes (6%) (Table 2). Among the unigenes categorized into specific COG functional annotations, 4933 unigenes (6%) were categorized under “function unknown”.
3.3. Metabolic Pathway Analysis
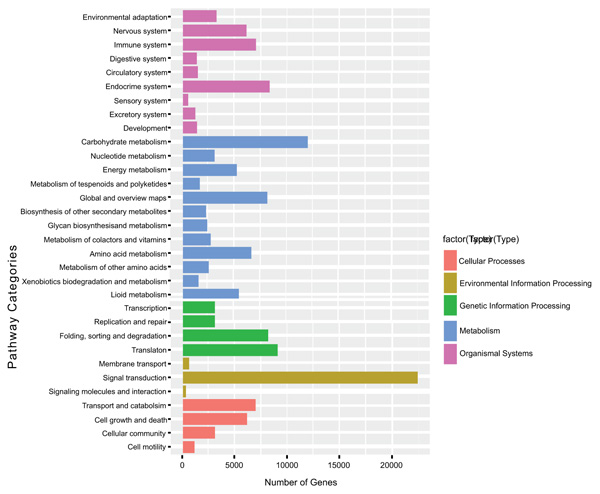
To understand the biological functions of the transcriptome genes on the biochemical pathways KEGG functional classification was performed and obtained 80,587 (19.8%) unigenes involved into 32 KEGG pathway categories. Among the KEGG pathways categories, signal transduction is the most represented with 25,000 genes (31%) followed by carbohydrate metabolism 12,000 genes (14.9%). The other pathway categories with a significant number of unigenes include Endocrine system, global and overview maps, translation, folding, sorting, and degradation pathway categories. These pathways have genes ranging from 8,000 to 9,000 genes (Fig. 4).
| Sequenced reads | Number of raw reads | 809,219,680 |
| Number of clean reads | 754,444,646 | |
| Total read length (bp) | 99,601,349,229 | |
| GC content | 53.72 | |
| Q20 percentage | 97.4 | |
| Contigs | Q30 percentage | 93.09 |
| Total number | 9,595,574 | |
| Total length(bp) | 1,101,480,760 | |
| Mean length(bp) | 374 | |
| Contig N50 (bp) | 566 | |
| Unigenes/transcripts | Total number | 406,181 |
| Total length (bp) | 240,231,095 | |
| Mean Length(bp) | 641 | |
| Unigene N50(bp) | 873 | |
| Minimum length(bp) | 201 | |
| Maximum length(bp) | 16651 |
| Code | Description | Gene_num |
|---|---|---|
| Metabolism | ||
| C | Energy production and conversion | 3803 |
| Q | Secondary metabolites biosynthesis, transport and catabolism | 2833 |
| P | Inorganic ion transport and metabolism | 2480 |
| F | Nucleotide transport and metabolism | 1072 |
| I | Lipid transport and metabolism | 3430 |
| H | Coenzyme transport and metabolism | 980 |
| G | Carbohydrate transport and metabolism | 4848 |
| E | Amino acid transport and metabolism | 4136 |
| Information Storage and Processing | ||
| L | Replication, recombination and repair | 2240 |
| B | Chromatin structure and dynamics | 1179 |
| A | RNA processing and modification | 3702 |
| K | Transcription | 4928 |
| J | Translation, ribosomal structure and biogenesis | 4987 |
| Cellular Processing and Signaling | ||
| O | Posttranslational modification, protein turnover, chaperones | 10191 |
| W | Extracellular structures | 266 |
| Z | Cytoskeleton | 2343 |
| M | Cell wall/membrane/envelope biogenesis | 1328 |
| N | Cell motility | 38 |
| D | Cell cycle control, cell division, chromosome partitioning | 2066 |
| T | Signal transduction mechanisms | 12861 |
| Y | Nuclear structure | 250 |
| V | Defense mechanisms | 722 |
| U | Intracellular trafficking, secretion, and vesicular transport | 4968 |
| Poorly characterized | ||
| S | Function unknown | 4933 |
| R | General function prediction only | 9993 |
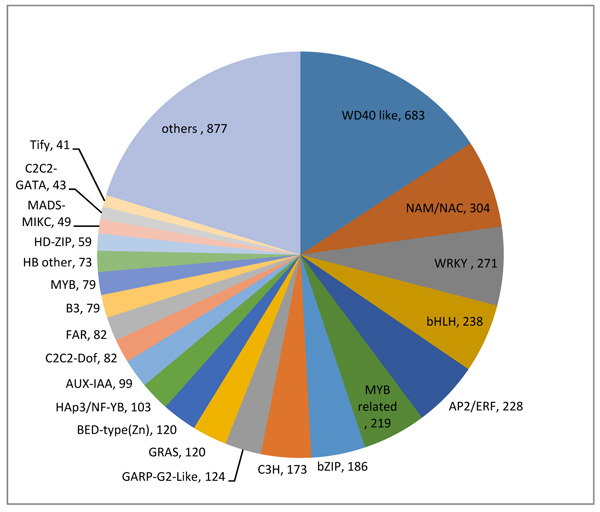
3.4. Prediction of Transcription Factors and Protein Family Assignment
Transcript factors have been exhibited to engage in key tasks in reaction to abiotic stresses by regulating gene expression [28, 29]. For a comprehensive understanding of O. abyssinica’s gene control and regulation, transcript factors were predicted using family assignment rules established at http://itak.feilab.net/cgi-bin/itak/online_itak.cgi [30]. According to family assignment rules 4,332 Transcript Factors (TFs) were predicted to have active involvement in the regulations of gene expression and these TFs were organized into 64 transcription factors protein families. WD40, NAM/NAC, WRKY, bHLH, AP2/ERF, MYB relate and bZIP, were the most dominant TFs families (Fig. 5). These sequences specific DNA-binding proteins are believed to take part in a decisive role in stress signal transduction pathways. TFs families with proven roles in the regulation of stress response in plants include, NAC, WRKY, AP2/ERF, bZIP, and MYB related [31]. In this study the most represented TF families with differentially expressed genes (DEGs) includes, bZIP (49), WRKY (43), MYB (38), AP2/ERF (30), HD-ZIP (25) and MYB related (21), which demonstrates members of these gene families are closely associated with abiotic stress defense [32, 33].
3.5. Differential Expression Genes (DEGs) Analysis
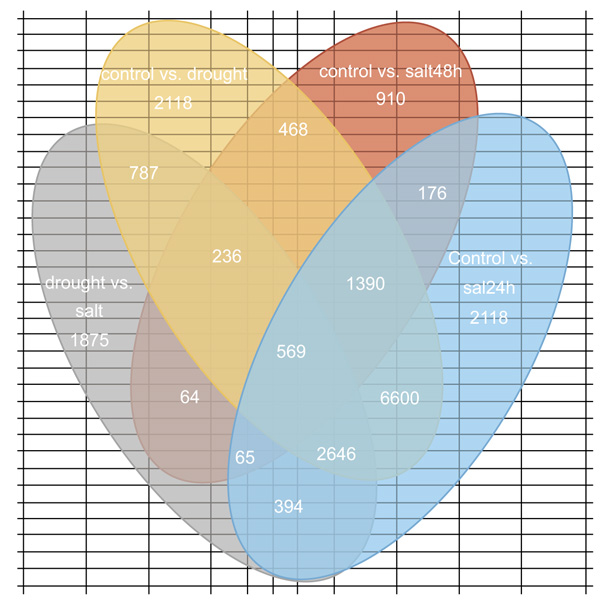
The direct expression of a gene's expression level is the abundance of its transcript, the higher the degree of transcript abundance, the higher the gene expression level. DEGs analysis used three combinations: (i) Control vs. drought, (ii) control vs. salt and (iii) drought vs. salt. The number of DEGs common and specific between the different stresses were presented in Fig. (6). Clustering analysis was conducted to: (i) calculate the similarity of the data and classify the data according to the similarity so as to cluster together the genes with the same function or close relationship, (ii) to identify the unknown gene function or the unknown function of the known gene and (iii) to infer whether they commonly participate in the same metabolic process or cell pathways.
Differential Expression Genes (DEGs) analysis from this abiotic stress-induced transcriptome study resulted into the expression 65,471 stress-responsive genes, 29,746 up-regulated and 35,725 down-regulated. This indicated that these DEGs are involved in regulating the response to drought and salt stresses. Due to drought stress, a total of 34,719 DEGs were identified, among these 15,681 were up-regulated and 19,038 were down-regulated (Supplementary file 2). Out of 13958 salt 24h stress DEGs, 5994 up-regulated and 7964 down-regulated (Supplementary file 3). Out of 3878 salt 48 h DEGs, 1289 were up-regulated and 2589 were down-regulated (Supplementary file 4). Out of 12,916 droughts vs. salt DEGs, 6,782 were up-regulated and 6,134 were down-regulated (Supplementary file 5). Although drought stress has one technical replication the numbers of its DEGs are almost double compared to salt stresses which have two technical replications. This suggests that O. abyssinica is more responsive to drought than salt stress. In both drought and salt stresses when compared to control, the numbers of down-regulated DEGs are greater than up-regulated ones. But in the case of drought vs. salt DEGs analysis, up-regulated genes are greater. When we observe DEGs among treatments, genes co-expressed exclusively in each stress includes 15,745 for control vs. drought, 2,118 for control vs. salt 24 h, 910 for control vs. salt 48 h and 1,875 for salt vs. drought. But 25,150 DEGs were shared by two or more groups of treatments while 569 were shared by all stress (Fig. 6). DEGs shared by all stresses groups might be blessed in conferring multiple stress tolerance so that those genes might serve as potential candidates for exploring multiple stress tolerance and adaptation.
Out of 65,471 DEGs, 14, 258 (21.8%) are functionally annotated. Most importantly 78.2% of the DEGs were not classified to any functional pathways, which suggests these putative novel genes might be species-specific and these genes might be involved in key pathways regulating in stress adaptation.
Out of 29,746 up-regulated genes, only 7,103 (23.8%) could be assigned to putative functions. Some genes have a wide range of functions by involving dozens of pathways. For instance, in drought stress, GO: 0000165, GO: 0005524, GO: 0080136, each involved in 97 pathways. In salt 24 h stress GO: 0080136 (97), GO: 0010099 (37), GO: 2000021 (23). In salt 48 h, GO: 0005739 involved in 18 pathways. In drought vs. salt, GO: 0080136 in 97 pathways.
Out of 35,725 down-regulated genes, only 7,155 (20%) were assigned to functions. The down-regulated genes with the most diverse functions by participating in dozens of pathways includes, in drought stress, GO:0000165, GO:0005634, GO:0042542 and GO:0005524 all under the term mitogen-activated protein kinase involves in 97 pathways, GO:0005952 in 63 pathways. In salt 24, GO: 0005952 (64), GO: 0005509 and GO: 0009536 each in 37 are with the most diverse functions. In salt48h, GO: 0004721 (21) are the most multi-functional genes. In drought vs. salt, GO: 0007264 (41).
3.6. Validation of RNA-Seq Data by qRT-PCR
In order to confirm the credibility of the RNA-Seq data, qRT-PCR was conducted. Randomly chosen 20 up-regulated and 20 down-regulated genes were used. To validate the expression levels measured by RNA-Seq, the ratio of expression of selected genes as measured by qRT-PCR was compared to the ratio of expression under drought and salt stress conditions using RNA-Seq. From 40 selected unigenes, only 13 unigenes could be quantified due to specificities of primers. But because of sample replications, these unigenes were quantified from 28 gene samples. The RNA-Seq data have strong positive correlation coefficient value with qRT-PCR, since the value of R2 = 0.90.
4. DISCUSSION
The average length of 641 bp, N50 of 873 bp was more or less comparable with other de novo transcriptome assemblies in other plant species [34-38]. The higher ploidy level (hexaploid) nature of O. abyssinica might be the reason behind 406,181 unigenes. On the contrary de novo assembled hexaploid hulless Oat has generated 128,414 putative unigenes [39], transcriptome analysis of five hexaploid and allododecaploid Spartina species generated unigenes ranging from 13,054 to 16,002 [40]. In the species similarity search, O. abyssinica showed less similarity with Moso bamboo. This genetic divergence might be due to the existences of more than 1,250 species of bamboos across the globe, differences in speciation time, variation in selection pressure and geographic distances.
When compared to transcriptome analysis of other plants, the 217,067 number of functionally annotated unigenes of O. abyssinica were much higher. Although the number is higher, the percentage of annotated unigenes is smaller (53.4%). For instance, the number of unigenes and annotated percentage of some species includes, 59,814 unigenes (73%) in drought and cold stresses response of Ammopiptanthus mongolicus, 177, 817 unigenes (94.8%) in salt response of Spartina alterniflora, 54,125 unigenes (58.7%) in desiccation-tolerant moss Syntrichia caninervis, 95,897 unigenes (83.57%) in Miscanthus sinensis [34, 35, 41, 42].
In O. abyssinica, of the total 406,181 unigenes, 217,067 unigenes were functionally annotated. The remaining 189,114 (46.6%) unigenes did not show any significant functional similarity to any of the database explored. This may be attributed to the lack of references genome of O. abyssinica and the highly divergent nature of the unigenes or novel genes that carry out species-specific functions. The GO analysis revealed that binding and catalytic activity from molecular function and metabolic process from the cellular component were represented with a high number of unigenes. More or less similar supporting results were found in transcriptome analysis of Ammopiptanthus mongolicus, Nitraria sibirica and Urochloa decumbens under abiotic stresses [35, 36, 38], this suggests that genes involved in the above GO terms are responsive to abiotic stress.
Only 20% of unigenes were assigned to COG database and the majority of these unigenes goes to cellular processing and signaling pathway categories. These two dominant subcategories were also with the highest unigenes in a transcriptome analysis of Moss, Tall Fescue and Tobacco [41, 43, 44]. This suggests large numbers of genes in cellular processing and signaling are actively involved in drought and salt stresses response.
Functionally unclassified 4933 unigenes might stand for lineage and/or species-specific genes for adoptive innovation and could be untranslated regions of the transcriptome or novel genes that perform species-specific functions. Signal transduction mechanism at COG and signal transduction at KEGG analysis are represented by the highest numbers of genes, which suggests that both the genes involved and the functional pathways actively respond to stress [35].
The metabolic pathways analysis result suggests that signal transduction and carbohydrate metabolism events are active in O. abyssinica response in the face of drought and salt stress. Similar supporting results were obtained from Cotton and Buckwheat transcriptome analysis under abiotic stress [39, 45].
TF families showing up-regulation for both stresses could be targets for studying plant response in the face of unfavorable environmental conditions. Whole genome expression profiling and transcriptome studies in many plants have confirmed the stress responsiveness of various TF families, C2H2 in Populus trichocarpa [46], NAC in Soybean [47], WD40 in Foxtail Millet [48], WRKY in carrot [44] and Broomcorn millet [49]. In addition to wild plants, many studies have been conducted on the role of TF families in stress response of transgenic plants, like AP2/ERF conferred stress tolerances in Populus simonii x populous nigra [50], manipulation of specific NAC TFs has conferred stress tolerance in transgenic Rice, Tobacco, Wheat and Arabidopsis [51], AP2/ERF in transgenic Trifolium alexandrinum [50], bHLH in Arabidopsis thailina [52, 53]. The involvements of TF families in diverse abiotic stresses have been reported in different plants [54-56].
Analysis of differentially expressed genes has revealed abiotic stress responsiveness of O. abyssinica genome. A total of 65,471 DEGs from these 29,746 up-regulated and 35, 725 were down-regulated. DEGs analysis of hexaploid hulless oat has generated 65,000 unigenes [39]. The functionally annotated 14,258 genes proved to be actively involved in diverse pathways activities. As many genes involved in similar functions, the most represented proteins by up-regulated genes include, heat shock 70kDa protein 1/8, serine/threonine-protein kinase SRK2, protein phosphatase 2C, beta-glucosidase, glutathione S-transferase protein xylosyltransferase, GTP-binding nuclear protein Ran, CTP synthase, adenylate kinase, Delta 1-pyrroline-5-carboxylate Synthetase, glutamine synthetase, Chitinase and glyceraldehyde-3-phosphate dehydrogenase. Majority of the above proteins take part in an important role in the regulation of stress tolerance. Serine-threonine protein kinases are proven to take part in the regulation of signaling cascades and some of these when over-expressed improved stress-tolerance of plants [57]. Glutamine synthetase involves incorporating toxic free ammonium ions into glutamate and glutamine, respectively; thus up-regulation of these genes may be a possible mechanism of stress tolerance [58]. Largely represented up-regulated genes like Delta 1-pyrroline-5-carboxylate Synthetase, glyceraldehyde-3-phosphate dehydrogenase and Chitinase play important role in plant defense and enhance resistance and tolerance to drought and salt stresses [35, 59].
The most prevalent proteins encoded from down-regulated DEGs includes, interleukin-1 receptor-associated kinase 4, aldehyde dehydrogenase (NAD+), glutamine synthetase, fructose-bisphosphate aldolase, phosphoenolpyruvate carboxylase, cysteine synthase A, (S)-2-hydroxy-acid oxidase, glutamate synthase (ferredoxin), thioredoxin reductase (NADPH) and light-harvesting complex II chlorophyll a/b binding protein.
Among the down-regulated genes, with a significant role in stress tolerances were GABA transporter, arginine, which is one of the precursors of putrescine, glutamate which is the precursor molecule for proline (an osmo-protectant) plays important role in plant stress-regulation and stress-tolerance [60]. Phosphatase 2C family protein also played a significant role in stress tolerance [61]. Receptor protein kinase-like and Glucose-6-phosphate dehydrogenase are also believed to confer stress tolerances in Barley dehydration shock and drought stress [62].
CONCLUSION
This study has investigated and presented the first comprehensive global transcriptome profiling of O. abyssinica under drought and salt stress conditions using the Illumina sequencing platform. The study showed that multiple pathways are involved in drought and salt tolerance. Differentially expressed genes analyses have suggested the stress responsiveness of O. abyssinica transcriptome to drought and salt stress since 65,471 genes were differentially expressed. The predicted 4,332 Transcriptions Factors (TFs) organized into 64 TFs families uncovered the most important protein families associated with abiotic stress response. The commonly regulated 569 genes in both abiotic stresses are potential candidates for engineering multiple stress tolerance and adaptation in plants. In general, this transcriptome analysis had identified the key genes, transcription factor families, pathways that can be easily exploited for further transgenesis experiments aiming to investigate genes that are blessed in conferring stress tolerance and adaptation in plants.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
We are so much grateful to State Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University, Harbin China, for funding the study.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We would like to thank Ethiopian Biodiversity Institute for providing O. abyssinica seeds used for this RNA-Seq study.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
Supplementary file 1: Primers used for qRT-PCR validation of RNA-Seq data
Supplementary file 2: DEGs analysis for drought stress
Supplementary file 3: DEGs analysis for salt24 h
Supplementary file 4: DEGs analysis for salt 48h
Supplementary file 5: DEGs analysis for drought vs. salt

