All published articles of this journal are available on ScienceDirect.

Cellulolytic Activities of the Dung Beetle, Euoniticellus Intermedius, Larva Gut Micro-Flora
Abstract
Background:
The life style and biology of dung beetles offer a significant opportunity for innovation in biofuel production. The larvae of the African dung beetle, Euoniticellus intermedius, feed solely on cow dung, eating and digesting the fibre while adults live on juices found in fresh dung. The larval gut system consists of a small, almost unrecognizable foregut and two distinct chambers; the midgut and hindgut. It is clear that these two chambers are the centres in which the dung material whose composition includes cellulose is processed. The goal of this study was to assess the cellulolytic activities of cultured gut micro-flora derived from E. intermedius, (Coleoptera: Scarabaeida).
Method:
Late second to third instar stage E. intermedius larvae were dissected and the isolated gut micro-flora consortia aerobically cultured in media containing cellulose (filter paper) as the sole carbon source. Genomic DNA isolation was done on the gut consortia cultures after 10 days of culturing, using the ZR Fungal/Bacterial DNA MiniPrep kit (Zymo Research, USA). A complete and unbiased primary cosmid library was then constructed from the isolated genomic DNA using a cloning ready, pWEB-TNC™ Cosmid Cloning kit (EPICENTRE Biotechnologies, USA). The primary cosmid library clones were screened for endo-glucanase and cellobiohydrolase activities using Carboxymethyl Cellulose (CMC) and 4-Methylumbelliferyl-β-D-Cellobioside (MUC) plate assays respectively.
Results:
Results indicate that a total of 7 colonies out of 160 screened colonies showed positive CMC and MUC activities.
Conclusion:
This proves that E. intermedius is a potential source of cellulolytic micro-organisms and enzymes that can be used for cellulose derived biofuel production.
1. INTRODUCTION
The world’s dependence on fossil fuels to power our domestic, industrial and transportation energy requirements has been cited as one of the main factors that aggravate the perils of global warming and climate change by releasing greenhouse gases such as Carbon Dioxide (CO2) into the atmosphere [1, 2]. The effects of climate change are no longer a myth, since we are already experiencing them. Global warming and climate change have damaging environmental consequences and pose disastrous global risks for life on earth [3]. The need to alleviate the consequences associated with rampant fossil fuel use has seen an upsurge in investments in renewable and sustainable energy resources. Production of first generation bio-fuels has been well documented, but due to the food versus fuel controversy and the pressure put on the few available food resources, second generation bio-fuels have entered the fray as attractive alternatives. Despite offering high sustainability hopes, production of second-generation bio-fuels from lignocellulose biomass is still hampered by the recalcitrant nature of lignocellulose matter [4].
To tackle these challenges, chemical and biochemical methods for cellulose degradation to form reducing sugars and the subsequent fermentation of these reducing sugars to bio-ethanol have been extensively studied and proposed. On the biological front, various cellulose-degrading micro-organisms have been discovered through genomic and metagenomic approaches and the potential role of these micro-organisms in advancing the bio-fuel dream can never be over-emphasized. Research by many scientists has shown that cellulases exist in various natural environments such as water and soil, and are also produced by a wide range of organisms such as microbes, plants, insect and animals [5-8]. The digestive tracts of some insects reportedly harbour symbiotic microorganisms that help the hosts to digest the cellulosic feed [9]. Cellulose digestion has been specifically demonstrated in representatives of widely different taxonomic groups of insects: cockroaches and various wood-eating insects, lower and higher termites (Isoptera), various beetles (Coleoptera and wood wasps (Hymenoptera) [10]. This study was conducted to assess the cellulolytic activities of the dung beetle, Euoniticellus intermedius, and their potential application in the production of cellulose derived bio-fuels and other bio-products.
2. MATERIALS AND METHODS
2.1. Dung Beetle Larvae Dissection
Late second to third instar larvae were dissected according to a modified dung beetle dissection protocol similar to the one described by Lemke and colleagues [11]. The steel/metal dissection equipment was autoclaved at 121°C for 15 minutes. Insect ringer solution was prepared according to the protocol described by Hayashi and Kamimura [12] and autoclaved for 15 minutes at 121ºC. Larvae dissection was performed in a sterile preparation dish under a dissecting microscope, in a laminar flow cabinet. The larvae were first anesthetized by exposing them to a nitrogen, hydrogen and carbon dioxide (71/7/22 vol/vol respectively) (Afrox grade) gas mixture for 15 minutes. They were then fixed onto the preparation dish with steel pins, with the larvae laid on its sides, and sterile insect ringer solution added. The cuticle was cut along the side lines and the ventral integument, circular muscles and trachea were then carefully removed. After removing the head from the larva, a circular cut was made on the anus to allow careful retrieval of the intestinal tract. The guts were then stored in 1.5 ml tubes at -70°C.
2.2. Culturing of Dung Beetle Larvae Gut Micro-Flora
2.2.1. Media Preparation
A basal cellulose medium for E. intermedius larvae gut micro-flora cultivation was prepared using cellulose (Whitman filter paper) as the sole carbon source, according to a modified method described by Lemke and co-workers [11]. To the basal media preparation, 1 ml of trace element solution (SL11) and 1 ml of selenium tungstate solution were added and then adjusted to pH 8 using a 0.1 M sodium phosphate buffer before autoclaving.
2.2.2. Media Inoculation
Larval guts weighing 1 g were homogenized in 9 ml of phosphate buffered saline (pH 7.4). The homogenate (1 ml) was then inoculated into 9 ml of cellulose media (in the place of Whitman filter paper, 0.405 g of cellulose microcrystalline was used during pre-culturing experiments). The inoculations were done in triplicate for each respective medium. The pre-cultures were incubated at 30°C for 48 hours in the dark, shaking at a speed of 80 revolutions per minute on a rotary shaker. Microbial growth was visually observed by the degree of turbidity of cultures after 24 hours and 48 hours of incubation period.
Inoculation of pre-culture samples (10 ml) into the main cultures (90 ml) was done in triplicate after 48 hours of pre-culturing. The main cultures were incubated at 30°C in the dark, shaking at 80 rpm.
2.3. DNA Isolation
Samples for DNA extraction were taken after 10 days of culturing in the main cultures. Genomic DNA isolation was done on cultured samples using the ZR Fungal/Bacterial DNA MiniPrep kit (Zymo Research, USA). About 1 ml of culture samples were transferred into 1.5 ml tubes and centrifuged at 13,000 x g for 5 minutes at room temperature. The supernatant was discarded and the pelleted cells from 4 or 5 tubes pooled together (to achieve approximately 20 mg of sample weight) to perform one mini-prep DNA isolation. DNA isolation was performed on the pooled cells according to the procedure described in the ZR Fungal/Bacterial DNA MiniPrep kit (Zymo Research, USA). The DNA isolated from the different replicate tubes was pooled together, concentrated by precipitation in ethanol acetate (24:1) and re-suspended in 100μl TE buffer. The isolated DNA (5μl) was mixed with 1μl of gel loading buffer (Blue) and ran at 100V for 45 minutes on 1% agarose gel in 0.5X TAE buffer. A GeneRulerTM 100bp DNA ladder (Fermentas, South Africa) (5μl) was ran alongside the DNA samples and the rate of migration was visualized using UV light. The genomic DNA concentration was determined using a Nano-Drop spectrophotometer (ND 1000). The isolated genomic DNA was kept at -20°C until further analyses.
2.4. Cosmid Library Construction
A complete and unbiased primary cosmid library was constructed from the isolated genomic DNA using a cloning ready, pWEB-TNC™ Cosmid Cloning Kit (EPICENTRE Biotechnologies, USA).
2.4.1. Insert DNA Preparation
Previously isolated genomic DNA suspended in TE buffer (41.2077ug) was first sheared by expelling it twice from a syringe through a small-bore needle, as described in the kit instruction manual. Shearing was done to generate random DNA fragments (30-45 kb). The size distribution of the DNA molecules were then examined by running a 5μl aliquot of genomic DNA on 0.8% agarose gel in 1X TAE buffer using 100ng of the T7, cosmid control DNA as a size marker.
The DNA was then end-repaired, as described in the kit manual, to generate blunt ends that allow cloning of the DNA into the prepared pWEB-TNC vector. The 80μl end-repair reaction mixture comprised of 10X End-Repair Buffer (8μl), 2.5mM dNTP Mix (8μl), 10mM ATP (8μl), nuclease free water (2μl), genomic DNA (50μl, which translates to approximately 20.6μg) and End-Repair Enzyme Mix (4μl).
2.4.2. Insert DNA Size Selection
The end-repaired genomic DNA (20-45kb) was size selected by running a 5μl aliquot on 1% low melting point agarose gel in 1X TAE buffer at 30V overnight, for approximately 12-14 hours. No ethidium bromide solution was added to both the agarose gel preparation and the running buffer. The T7 cosmid control DNA (100ng) was loaded as a size marker on each of the outer lanes and the end-repaired DNA between these two marker lanes. Following completion of gel electrophoresis, the two outer lanes containing the cosmid control DNA were carefully cut off from the gel, stained with ethidium bromide and viewed under UV light. The position of size markers on the cut outer lanes of the gel was noted using a pipette tip and the gel re-assembled to establish the position of the insert DNA on the unstained gel slice. Using a surgical blade, a 3mm wide gel slice containing insert DNA migrating between the size markers was cut and stored in 1.5 ml tubes at 4°C until further experiments.
2.4.3. In-gel Ligation Reaction
The gel slice containing the end-repaired and size-selected DNA was melted and in-gel ligation done according to the pWEB-TNCTM Cosmid Cloning kit instruction manual. The reaction was stopped before the In Vitro Packaging step, and the reaction mixture stored at -20°C overnight.
2.4.4. In Vitro Packaging (Using MaxPlax Packaging Extracts)
This was done according to the pWEB-TNCTMCosmid Cloning kit instruction manual. A single packaging extract was thawed and 25μl of the extract added to a 1.5 ml tube. The ligated cosmid DNA (10μl) was added to the tube containing the extract, mixed by pipetting and returned to the bottom of the tube by centrifugation for one minute at 10,000 x g. The reaction mixture was incubated at 30°C for 90 minutes, after which another 25μl of packaging extract was added and the reaction mixture re-incubated at 30°C for another 90 minutes. Phage dilution buffer (500μl) was added, followed by 25μl of chloroform and the reaction mixed by gentle vortexing after each addition. This was stored at 4°C until further experiments.
The titer of the packaged cosmids was determined by adding 10μl of the packaged cosmids to 100μl of previously prepared EPI100-T1R host cells. The reaction mixture was incubated at 37°C for 20 minutes to allow bacterial transformation. The infected bacteria (110μl) were then spread on LB-ampicillin selection agar plates (90 mm). The inoculated LB-ampicillin plates were incubated in the dark at 37°C overnight (for 12-16 hours). After incubation, colony growth was determined and the titer calculated as follows:

2.4.5. Amplification of Cosmid Library
The cosmid library was amplified as described by the pWEB-TNCTMCosmid Cloning kit instruction manual. An overnight culture of EPI100-T1R cells (100μl) was inoculated with 10μl of the packaged phages. This was incubated in the dark at 37°C, shaking at 100 rpm for 20 minutes. After that, 0.5 ml of LB broth was then added to the infected culture and incubated at 37°C for 45 minutes. The infected culture (600μl) was added and spread on 4 LB-ampicillin agar plates (90 mm). The plates were incubated upside down in the dark at 37°C overnight, for approximately 12-14 hours (until colonies, 0.2-0.3 mm in diameter, were noticed). The colonies were transferred to a sterile tube by adding 10 ml of LB broth and scrapping them off from the plates using an inoculation loop. The LB broth suspended colonies were then vortex mixed to disrupt any bacterial clumps and sterile glycerol added to a final concentration of 15%. The bacteria were then stored at -70°C in 500μl aliquots.
2.5. Cellulolytic Activity Plate Assays
Plate assays were used to screen for cellulolytic activities of the primary cosmid library. The cellulase activities that were investigated are endo-glucanase activity and cellobiohyrolase activity. No β-glucosidase activity assay was done.
2.5.1. Endo-Glucanase Activity Assay
Endo-glucanase plate assays were performed according to a modified method described by Teather and Wood [13]. Colonies of the primary cosmid library were replica-plated on LB-ampicillin plates (90 mm) and incubated in the dark at 37°C for approximately 12-14 hours. A Carboxymethyl Cellulose (CMC) overlay supplemented with ampicillin was prepared. In the place of IPTG, 1μl of 0.1 M lactose (Merk) was added to 1 ml of the CMC-ampicillin overlay media.
The CMC-ampicillin/lactose overlay mixture (4-8 ml) was added to the previously incubated LB-ampicillin agar plates and these were further incubated at 37°C for 12-16 hours, followed by staining with Congo red dye (1 mg/ml) for 15 minutes. The plates were then flooded with 1 M NaCl and left at room temperature for another 15 minutes. The appearance of a clear yellowish halo on a red background around the colonies was used to determine endo-glucanase activity. After suspected endo-glucanase activity was observed, hydrochloric acid (1 M) was then added to inhibit further enzyme activity. Hydrochloric acid also aids contrast and stabilise the visualized zones, by changing the dye colour from red to blue in the process.
2.5.2. Cellobiohydrolase Activity Assay
Cellobiohydrolase plate assays were done according to a slightly modified method described by Reinhold-Hurek et al. [14]. Colonies that had previously shown positive endo-glucanase activity were replica-plated on LB-ampicillin agar plates (90 mm) and incubated at 37°C for 14 hours. A 4-Methylumbelliferyl-β-D-Cellobioside (MUC) overlay supplemented with ampicillin and lactose was prepared as described for CMC overlay media above. The MUC overlay (4-8 ml) was then added to the LB-ampicillin agar plates and the plates were further incubated for 12-16 hours. Plates were then exposed to a 302 nm UV Transluminator and active colonies were identified by the appearance of a blue fluorescence.
3. RESULTS AND DISCUSSION
The primary cosmid library exhibited both endo-glucanase and cellobiohydrolase cellulolytic activities. Positive endo-glucanase activity was observed in 3 colonies out of a total of 77 screened colonies from 10 different LB-ampicillin agar plates, by the appearance of a clear halo around the colony on a blue background as shown in Fig. (2). Cellobiohydrolase activity was also observed in at least 4 colonies out of a total of 83 colonies screened for this activity. Positive colonies exhibited a blue fluorescence when exposed to a 302 nm UV radiation on a UV Transilluminator, as shown in Fig. (3).
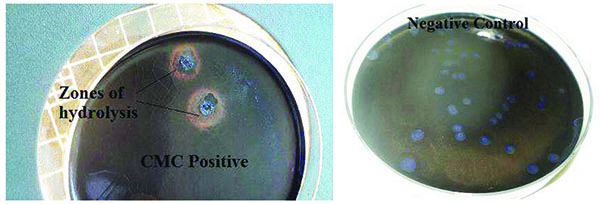
Endo-glucanase activity was evaluated using CMC as the cellulose substrate and the plates stained with Congo red dye to check for colony clearing zones. Endo-glucanases have been reported to be very active on the amorphous regions of cellulose and as such their activity are easily assayed using soluble cellulose substrates like CMC [15]. According to Teather and Wood [13], Congo red is a pH dependent dye that binds the β-1,4-glucosidic bonds of cellulose. Endo-glucanases, randomly cleave these β-1,4-glucosidic bonds along the cellulose chain and in the process releases the Congo red dye. This leads to the formation of a clearing zone or halo around the endo-glucanase positive clones. Cellobiohydrolase activity, on the other hand, was evaluated using MUC as the cellulose substrate source. This type of cellulase is exo-acting, as it cleaves β-1,4-glucosidic bonds from the cellulose chain ends [15]. Positive MUC activity was evaluated by the appearance of blue fluorescing colonies when the incubated plates were exposed to 302 nm UV light. The use of fluorometric MUC cellulose substrate in this assay allowed the detection of cellobiohydrolase positive clones [16]. It should be noted that the clones which had previously tested positive for endo-glucanase activity were replica-plated on LB-ampicillin agar containing MUC substrate and tested for cellobiohydrolase activity. Therefore, colonies that tested positive for cellobiohydrolase activity possessed both endo-glucanase and cellobiohydrolase activities. This is in agreement with Karmakar and Ray [15]’s previous claim there is evidence that some cellulases possess both endo- and exo- cellulase activity.
These positive cellulolytic activities observed confirmed previous research findings of many scientists who postulated that scarab beetle larvae had cellulose degradation capabilities due to the existence of a symbiotic relationship with their cellulolytic gut micro-flora [17-19]. The dung beetle gut bacterial symbionts provide the host with a broad array of metabolic activities and this significantly expand the dung beetle’s digestion spectrum through the degradation and fermentation of cellulose and other non-digestible plant fiber components [20].

Dung beetles belong to the family Scarabaeidae and order Coleoptera, which is regarded as the largest order in the insect kingdom. Scarab beetles are commonly found in various environmental niches where decaying plant and/or animal wastes form a high proportion of the available biomass [21]. This is because scarabaeids are reportedly herbivorous or saprophagous and many of their species feed on plant roots, decaying organic matter and animal waste of very low nutritional value [19]. As such, scarab beetles play an important role in the ecological control of dung, decaying wood and plant matter [22].
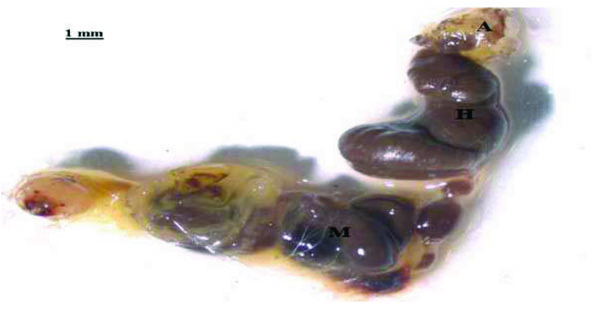
The gut system of E. intermedius larvae consists of two distinct compartments; a long cylindrical midgut and a bulbous hindgut shown in Fig. (1). It resembles that of humus-feeding larva of Pachnoda ephippiata (Coleoptera: Scarabaeidae) as reported by Lemke et al. [11]. The gut comprises of a rich population of xylan and pectin degrading micro-organisms, which increase in density from the mid-gut to the hindgut [17]. Cellulose and hemicellulose digestion in the scarab beetle larvae is thought to occur in the hindgut (H) section, aided by the symbiotic micro-flora [19]. The scarab hindgut is closely analogous to the rumen of higher mammals, which is the primary site of microbial fermentation of plant organic material [21]. The absence of cellulose-degrading micro-organisms and the non-reducing conditions in the mid-gut makes cellulose digestion in this gut compartment very unlikely. Thus, the mid-gut (M), (Fig. 1), only serves to prepare the cellulose or hemicellulose matter for further hindgut cellulose digestion [21].
There are two main methods currently being employed for characterizing gut micro-flora and these are classified as culture dependent and culture independent approaches. To date, most microbial products have been obtained from microbes that have been isolated and exploited in the laboratory [23]. However, advances in the field of metagenomics have dramatically revised our view of microbial biodiversity and its potential for biotechnological applications. It has been widely estimated that up to 99% of microorganisms present in most natural environments are non-culturable, and as a result, very little is known about their genomes, genes and encoded enzymatic activities [23-26]. While the culture independent methods are able to detect less frequent species, especially, those that cannot be cultured, the laboratory enrichment methods used in this study, although labour intensive, have been reported to be highly efficient for the rapid isolation of large DNA fragments and cloning of genes with high biotechnological value [26]. In addition the isolated bacteria can be further functionally characterized using traditional microbiology tools and are readily available for potential biotechnological applications. This strategy is commonly used in to increase the likelihood of detecting clones displaying the desired trait [27].
CONCLUSION
Whilst a number of studies had been done on scarab beetle larvae such as C. zealandica, P. ephippiata and Melolontha melolontha [21], no previous work has been previously documented to investigate E. intermedius gut consortia’s cellulose degradation capabilities, thus making this work a first. With the successful culturing and cellulose screening of micro-flora possessing cellulolytic activities from this dung beetle, a new source of cellulolytic biocatalysts has been established that can find wide application in the biomass derived biofuels and biotechnology industry.
LIST OF ABBREVIATIONS
| CMC | Carboxymethyl Cellulose |
| DNA | Deoxyribose Nucleic Acid |
| MUC | Methylumbelliferyl-β-D-Cellobioside |
| UV | Ultra-Violet |
| TE buffer | Tris EDTA buffer |
| TAE buffer | Tris Acetate EDTA buffer |
| LB | Luria Broth |
| rpm | revolutions per minute |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are base of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The author declares no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
I would like to thank my colleagues at Great Zimbabwe University, Dr. Washington Mutatu and Mr. Lazarus Chapungu for taking time out of their busy schedule to assist in editing this manuscript. Special thanks are due to Dr Karl Rumbold, Dr Monde Ntwasa and Mr. Phanaknosi Moyo for laying the foundation upon which this study was established. Lastly, this study could not have been successful without unconditional support from my friends and family.

