All published articles of this journal are available on ScienceDirect.

The Role of Endocrine Disruptors on Metabolic Dysfunction
Authors Info & Affiliations
Abstract
Abdominal obesity appears to be an important component of the metabolic syndrome (MetS), in which along with insulin resistance, hypertension and dyslipidaemia represents an increased risk for developing cardiovascular diseases and type 2 diabetes (T2D). The aetiology of obesity and its comorbidities is multifactorial, but despite the evidence of traditional contributing factors, the role of environmental toxicants with endocrine disrupting activity has been recently highlighted. Indeed, even small concentrations of these endocrine disrupting chemicals (EDCs) have the ability to cause severe health damages. In this revision, we focused our attention on the mechanisms of action and impact of EDCs exposure as a contributor to the present epidemics of obesity and MetS.
The "environmental obesogens" hypothesis associates environmental EDCs to the disruption of energy homeostasis, with recent studies demonstrating the ability of these compounds to modulate the adipocyte biology. On the other hand, the distinct distribution pattern observed between two metabolically distinct AT depots (visceral and subcutaneous) and subsequent repercussion in the aggravation of metabolic dysfunction in a context of obesity, provides accumulating evidence to hypothesise that EDCs might have an important “environmental dysmetabolism” effect.
However, in addition to adulthood exposure, the perinatal effects are very important, since it may allow a change in the metabolic programming, promoting the further development of obesity and MetS. Therefore, additional research directed at understanding the nature and action of EDCs will illuminate the connection between health and the environment and the possible effects triggered by these compounds in respect to public health.
INTRODUCTION
Worldwide, cardiovascular diseases (CVD) continue to be the major cause of mortality and morbidity, with its incidence increasing alarmingly in the developing world. Type 2 diabetes (T2D) is rising in tandem due to increasing obesity and decreasing physical activity, also fuelling the increase in CVD [1]. The term “metabolic syndrome” (MetS) denotes the clustering of risk factors for T2D and CVD, and its reduction has become one of the major public health challenges worldwide, as it is mentioned on top of the 21st century civilization diseases list [2-5]. This condition is characterized by the manifestation of at least three of the following components: high blood pressure, low HDL-cholesterol, high triacylglycerols, high fasting glucose and abdominal obesity [6]. The syndrome is particularly useful in a practical clinical sense, drawing attention to those at increased risk of CVD and T2D and allowing early intervention [1]. Due to its close relationship with all the others components, some authors consider abdominal obesity as determinant in the causal pathway of the MetS, impacting on the other features and on CVD. In fact, obesity and in particular visceral obesity is associated with dyslipidaemia and insulin resistance, and results from the most recent research do indeed support such a central and causal role of visceral adipose tissue (AT) in this ethiopathogeny, possibly as a result of the particular localization and unique metabolic characteristics of this AT [7].
The rise in obesity prevalence has been for some time a major global public health problem. Indeed, as our society changes, also the main concerns about human health evolve alongside with our lifestyle and the way we interact with the surrounding environment. What started being a concern of the wealthy nations, mainly due to their abundance based society, soon began to spread worldwide reaching epidemic proportions [8-10]. Adding to these concerns, the future does not seem promising as child excessive weight and obesity are also rapidly increasing, an alarming perspective as it foretells them as obese adults [11].
The genesis of obesity is multifactorial and although there are many theories about the causes of the obesity epidemic, to date, there is still much uncertainty about its aetiology. Nevertheless, in an ultimate analysis it is dependent on a chronic positive disruption of the body’s energy balance equation - energy intake versus expenditure, on a background of genetic predisposition - as the central driving mechanism that promotes obesity [12, 13]. Even though much research has focused on these factors, the short time in which this shift in obesity expansion occurred raised several contradictions [8]. Nonetheless, the feedback mechanisms and pathways of homeostasis control are somehow altered, fact that, in addition to the diversity in weight reduction responses and comorbidity manifestations, could indicate that still unidentified factors may also play a role in the disruption of body energy balance.
In this regard, emerging from the profound environmental changes triggered by man in the last century and the knowledge that environmental cues can not only influence the individual’s behaviour, but also have a direct biological effect on the regulation of energy homeostasis controlling mechanisms, a plausible but also provocative theory was proposed by Baillie-Hamilton, associating the exponential increase of chemical substances in the environment over the past 450 years with the exponential increase in obesity prevalence [8]. Indeed, this evident parallel increase suggests a possible causal link between the two occurrences. These pollutants, man made synthetic compounds that even though being initially designed for a specific industrial function or those related with the growth of industrialization and the use of pest control in agriculture, etc., are now being linked with a wide range of side effects as when absorbed into the body they can act in the disturbance of normal homeostasis. In fact, several of these chemical pollutants are known as endocrine disrupting chemicals (EDCs), since even at very low doses they alter endocrine signalling, mimicking or blocking endogenous hormones or modulating transcription factors, changing epigenetic regulation and endogenous hormone kinetics/availability, being proposed for a number of adverse human health effects, including infertility and cancer [14-17]. Their particular characteristics when present in very low concentrations are well as reflected in the unusual dose-effect curves, where we can observe U and inverted U-shaped curves contrasting with traditional linear toxic effects. For these reasons, the analysis and regulatory decisions on EDCs should be based on the principles of endocrinology, and not only under a classical toxicological point of view [14, 15].
Moreover, because of the large variety of suspected EDCs, humans and animals are most likely exposed not to a single agent, but rather to a mixture of multiple EDCs [18]. So, the analysis of complex mixtures and of the body burden in EDCs are also a concern in actual toxicology and EDC research. Since the beginning of this concern, an international effort is being made to understand the true magnitude of human contamination, but despite the commitment of some nations with interesting monitoring programs, many others continue to disregard this hazard [19]. For this reason, the clear demonstration of EDC effects on human health as well as the development of easy and cost/effective procedures for their identification and quantification in the environment and biological samples is a determinant step for the enforcement of more and better monitoring programs and adds evidence for their wide recognition as health hazards.
EDCS EVALUATION IN HUMANS
Most EDCs are synthetic organic chemicals (xenobiotics) introduced into the environment by anthropogenic inputs [18]. A shared characteristic to all is that they contain at least one aromatic moiety in their molecular structure. Thus, their hydrophobic properties might consist of an important characteristic of their behaviour [18]. Depending on the EDC, their effects on biota have been observed at EDC concentrations as low as 0.1 ng/L [18]. The analysis of EDCs in human samples represents a difficult task because of the high complexity of the matrices analysed and because of the usually low concentration (ng/L) at which the target analytes are present in such samples. Human samples usually contain large amounts of possible interfering compounds that require the use of extensive extraction and clean up procedures to obtain extracts amenable to analysis [18]. Therefore, the development of sensitive and reliable techniques for the measurements of EDCS in order to assess the human exposure to EDCs and the possible correlated health effects is essential (Fig. 1). As a consequence, one of the major trends in analytical chemistry is the development of fast and efficient procedures for the trace analysis of target and non target organic compounds in complex matrices [18].
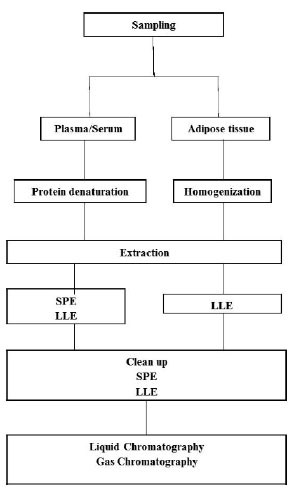
Scheme of analytical methods for the determination of EDCs (namely persistent organic pollutants) in most frequent analysed human samples (plasma and adipose tissue). LLE, liquid-liquid extraction; SPE, solid phase extraction.
Sample preparation and clean up are necessary to remove interferences that would otherwise affect the determination of the analytes; to enrich the target analytes; and to perform solvent changing to the desired solvent conditions used for instrumental detection [18]. Sampling and sample preparation are of crucial importance in an analytical procedure, representing quite frequently a source of errors [20]. For liquid samples, such as plasma/serum and urine, it is generally necessary to precipitate the protein content by adding acids, or organic solvents, or even applying ultrasound equipment or a combination of both [21, 22]. Solid samples, as the AT, require an adequate homogenization before sample extraction, normal with organic solvents applying a homogenizer. Generally, for the extraction of EDCs from liquid and solid samples such as plasma/serum and AT, liquid-liquid extraction (LLE) and solid phase extraction (SPE) are the most common techniques [18, 23].
LLE is a technique also known as solvent extraction and partitioning used to separate compounds based on their relative solubilities in two different immiscible liquids, usually aqueous samples and an organic solvent. It is an extraction of analyte from one liquid into another liquid phase [21]. SPE is a extraction technique that fractions the target analytes based on their affinity to the sorbent [24]. In this technique, in order to retain the studied analytes, one has to optimize the choice of the stationary phase, solvents (conditioning, washing and eluting), pH and flow rate. In other hand, the chosen SPE method should be able to remove as much as possible the matrix interferents [18, 25]. SPE, either alone or in combination with LLE, is one of most used techniques for the extraction from EDCs from plasma sample. For solid samples such as AT, samples are generally first extracted with organic solvents and then clean up through SPE technique [22, 23].
Fernandes et al. [23] analysed several organochlorine compounds in human AT by SPE-Gas Chromatography. Briefly, 0.2 g of AT was homogenized with 2 mL of hexane and 40 µg/L at high rpm using a homogenizer [23]. Then, 500 µL of the supernatant was transferred to a C18 cartridge. These cartridges were preconditioned with 1 mL of n-hexane under vacuum conditions, and after sample loading the samples were eluted with the same solvent and under vacuum. Then, the eluate obtained was evaporated to dryness, and the residue reconstituted in 1.0 mL n-hexane and analysed by GC-ECD and GC-MS. The SPE functioned in this work as a clean up step [23]. A similar technique was also applied in breast AT [26]. In this work, a different SPE clean up was used applying silica cartridges and final measurement by GC coupled to triple quadrupole MS [26]. Regarding plasma/serum samples, Covaci et al. [21] determined the presence of organochlorine pesticides in several human body fluids, including serum. Shortly, the procedure involved a denaturation of serum proteins with formic acid, SPE using C18 Empore™ cartridges, followed by elimination of lipids using cleanup on acidified silica or florisil [21].
Thus targeted analyses based on analytical standards provide the necessary sensitivity and selectivity, but lead to a fragmented picture of the occurrence of EDCs in humans. Consequently, there is a high demand for developing screening analytical methods that will accommodate a wide variety of analytical functional groups at low detection levels [27].
THE IMPACT OF EDCS ON HUMAN HEALTH
Over the past decades, there has been an increasing focus to investigate associations between human exposure to EDCs and disease [27], since due to their ubiquity in the environment and intrinsic properties, resulting in the contamination of virtually all individuals, they might have a substantial overall effect on the population [28]. Despite their hazards, these chemicals continue to be produced, used, and stored in many countries around the globe. Even where national bans or other controls exist, their restrictions are often poorly enforced and in any case, they cannot protect citizens from exposure to EDCs that have migrated from other regions where these chemicals are still in use. Even though some EDCs have already been banned in many countries. Their persistence is also related with the fact that production and release into the global environment still occurs in other countries [29].
Among these EDCs, attention was drawn to a specific group of man-made compounds, the persistent organic pollutants (POPs) [30]. As the designation suggests, they are extremely resistant to chemical and biological degradation that, allied with their lipophilic nature, contribute to their persistence in environment and consequent deleterious effects [31]. Being lipophilic, POPs are easily accumulated in fatty tissues (bioaccumulation) and their levels increase as we go up in the trophic chain (biomagnification). In everyday life, humans are mainly exposed through ingestion of fat-containing food such as dairy products, meat and fish, and, being at the top of the food chain, concentrate greater amounts of these compounds [31, 32]. It is not surprising that POPs can be detected in several human tissues and fluids, such as blood, breast milk and in the AT, their main reservoir [19, 28]. In addition to exposure via food and contaminated water, humans can also be exposed to these chemicals by inhalation of polluted air or cutaneous absorption via personal care products [19].
Another important group of EDCs are the plasticizers, such as phthalates and bisphenol-A, used in plastic materials, normally to increase their flexibility, as well as in cosmetic and pharmaceutical products [33-38]. As they do not usually accumulate in the body tissues and have very short half lives. Their metabolites are mainly detected in urine [39-41]. However, due to the widespread use of plastic there is a continued environment and human exposure to plasticizers compounds, reason why they are also considered “persistent” [33, 40].
Nevertheless, the true magnitude of human population exposure and its variation according to location in the globe is virtually unknown, complicating the definition of guidelines for research and health regulations. With the purpose to meet the community reserves regarding these threats, the United Nations Environment Program (UNEP) is raising a global awareness concerning the exposure to EDCs and their adverse outcomes, promoting worldwide research programs to determine the prevailing levels of EDCs in the population and to investigate health risk associated with background exposure [27, 30].
In search of relevant effects in humans, several authors found a relationship between exposure of EDCs in the organism and body weight increase through the disruption of normal energy homeostasis control, therefore beginning to designate these compounds as environmental “obesogens” [42]. Nevertheless, obesity itself is not the main source of concern but instead, the morbidities that arise associated with it [43]. Among them, the increase both in obesity and T2D are fuelling the maintenance of CVD as a major cause of global mortality [1]. More recently, a study from the American Diabetes Association also found a parallel increase of synthetic organic chemical production and T2D prevalence in the U.S. population between 1940 and 2010 [44]. Moreover, the involvement of the environmental chemicals in the development of obesity and metabolic disorders is now becoming an important area of discussion. Several studies have also begun associating the contamination of these compounds with metabolic dysfunction, interfering with the manifestation of other MetS components, namely dysglycaemia and dyslipidaemia [45].
Additionally, despite the paramount importance of evaluating the effects of individual EDCs in order to thoroughly understand their effects and mechanisms of action, one cannot disregard the fact that these compounds appear as complex mixtures in biological tissues which possibly influences their final effect [45]. Using cross sectional data from the 1999-2002 US National Health and Examination Survey, Duk-Hee Lee and colleagues reported a prevalence of a cluster of cardiovascular risk factors related to background exposure to a mixture of POPs, several of which also related to the prevalence of T2D. These findings raise the possibility that, in addition to obesity, some EDCs stored in AT may be a key to the pathogenesis of MetS [45].
The link between EDCs and MetS is reinforced by other studies such as the German Environmental Survey, the Arctic Monitoring and Assessment Program and population-wide studies in New Zealand, Australia, Japan, Flanders (Belgium) and Canary Islands (Spain). Nevertheless, few human biomonitoring reports and studies based on truly representative samples of the general population are available. The full outline of the distribution of EDC concentrations is unknown-if not ignored-in many countries worldwide. The available monitoring studies reveal the ubiquitous presence of these compounds in both blood and AT of individuals [32, 46-48]. On the other hand, the detection of EDCs in samples of umbilical cord blood, breast milk and semen raises issues are related to possible developmental effects [19].
Plasticizers, dioxins, polychlorinated biphenyls, dichlorodiphenyldichloroethylene (DDE, the main degradation product of the pesticide dichlorodiphenyltrichloroethane [DDT]), trans-nonachlor, hexachlorobenzene, and the hexachlorociclohexanes (including lindane) are some of the most commonly found EDCs in humans [28]. Although specific types of EDCs may have specific health effects. The synergistic and simultaneous exposure to several EDCs may lead to a range of health effects such as MetS [45]. Another debate is related with the effect concentration, since higher concentrations have a recognized toxic effect, unlike the lack of information for the lower concentrations that are far below the NOAEL (No Observed Adverse Effect Level) for these EDCs. The discovery that these lower concentrations, detected in human samples, may act on certain molecular mechanisms, leads to a reassessment of POPs effects, in order to identify: new obesogenic agents, possible molecular targets and potential cellular obesogenic mechanisms On the other hand, even among obese individuals one can observe a variation in the prevalence of cardiometabolic complication, where metabolically healthy subjects can account for 10-25% of the obese population [49]. In a recent paper, Gauthier et al. [50] confirmed a relationship of POPs with the variation in metabolic risk observed among obese individuals showing that the metabolically healthy but obese (MHO) phenotype is associated with lower plasma levels of POPs as compared with metabolically dysfunctional obese subjects. In fact, results obtained by our group studying 189 obese patients undergoing bariatric surgery revealed that, in addition to their ubiquity, POP levels either in subcutaneous (scAT) and visceral (vAT), the main reservoir of POPs, or their sum were higher in subjects with evidence of metabolic abnormalities, namely in glucose metabolism [51]. In our analysis, this pattern was especially evident for vAT, as we observed a dissimilar POP storage capability in the two distinct AT depots (vAT and scAT) with higher vAT POP levels in patients with increased aggregation of MetS components and higher 10-year cardiovascular risk based on the Framingham score. Hence, these chemicals may have a potential role in the development of metabolic dysfunction in a context of obesity, shifting the focus to their metabolic effects and broadening the view over their simple recognition as environmental obesogens subjects [51-53]. Additionally, AT POP levels seem to be associated with unsuccessful weight loss in obese patients after 12 months of surgery, as older individuals, who also had higher levels of AT POPs, lost less weight [51]. This suggests the possible involvement of POPs in this relationship and highlights their importance as negative markers of metabolic dysfunction reversibility in obese subjects.
Children are especially vulnerable to EDC exposure because they consume larger amounts of food and water relative to their body weight as compared with adults. Lopez-Espinosa et al. [54] conducted a study with the aim to investigate the presence of 16 organochlorine pesticide residues in 52 fat samples collected from boys with a mean age of 7 yr (0-15 yr) living in Southern Spain. No pesticide was found in more than 50% of samples, except for dichlorodiphenyltrichloroethylene (p,p'-DDE, 79% of samples; median, 710 ng/g lipid). Following this compound, the most frequent pesticides were dichlorodiphenyltrichloroethane (o,p′-DDT, 17%; median, 330 ng/g lipid) and dichlorodiphenyldichloroethane (o,p′-DDD, 15%; median, 1510 ng/g lipid) [54]. In another study Meza-Montenegro [55] assessed the exposure of children to different organochlorine pesticides, in major agricultural areas of Sonora, México. The study was undertaken in 165 children (age 6-12 years old) from 10 communities during 2009. Blood samples were analysed for organochlorine pesticides and all of the blood samples had detectable levels of p,p'-DDE, ranging from 0.25 to 10.3 ng/L. However, lindane, p,p'-DDT, aldrin, and endosulfan were detected in far less subjects (36.4, 23.6, 9.1, and 3%, respectively). Methoxychlor and endrin were not found in any sample. Therefore, further research is necessary to investigate the health consequence in children resulting from exposure to EDCs, as well as the mechanisms and molecular pathways involved in the disruption promoted by these compounds.
MECHANISMS OF EDCS
By definition, an EDC is a compound that alters the hormonal and homeostatic systems, the most investigated presently being related to sex hormones (estrogens and androgens, thyroid function and corticosteroids), although virtually any hormone system can be affected. De Coster and van Larebeke have reviewed in detail the mechanisms of action of EDCs [56]. These chemicals can act through nuclear receptors, nonnuclear steroid hormone receptors (such as the case of membrane estrogen receptors GPER [57]), nonsteroid receptors (for example, neurotransmitter receptors), orphan receptors (such as aryl hydrocarbon receptor, AhR), and also in pathways related to steroid biosynthesis and/or metabolism or on other actions that may impose onto the endocrine system.
Substances changing sex hormone signalling may have agonistic or antagonistic effects, i.e., they may be estrogenic, androgenic, antiestrogenic and antiandrogenic properties. This has been reported for compounds alone but also for complex mixtures of EDCs. Furthermore, substances may differ largely on agonistic/antagonistic potencies adding complexity to the study of their health effects [58]. EDCs such as bisphenol-A are known to act not only trough classic nuclear estrogen receptors (ERs), but also as potent estrogen via non-classical estrogen triggered pathways [59], leading to metabolic dysfunction and disruption of insulin signalling [60, 61]. Apart from exerting their effects though binding to receptors and changing transcriptional activity, some of these substances interfere with hormone synthesis as exemplified by the inhibition of aromatase [62]. Furthermore circulating EDCs may alter gonadotropin release and deregulate hypothalamus-pituitary-adrenal axis [63], with consequences not only to reproductive function but also to metabolism, given the known roles of sex hormones in the regulation of energy balance. Thyroid hormones regulate key steps in organ development (brain, ear, and bone) and on the metabolism of carbohydrates, lipids, and proteins, with impact on metabolic rate [64, 65], functions that become impaired upon exposure to EDCs with thyroid hormone-hormone disrupting abilities [56].
Another target of action is in the central mechanisms that coordinate the whole body’s response to daily nutritional fluctuations. The control exerted by the hypothalamic-pituitary-adrenal (HPA) axis plays a critical role to regulate energy homeostasis. In addition to systemic adrenal production, localized control over glucocorticoid signalling can be influenced by EDCs in peripheral tissues. Changes in 11β-hydroxysteroid dehydrogenase (11β-HSD) types 1 and 2 activities, the enzymes that mediate the inter conversion between cortisone (inactive) and cortisol (active) ligands respectively, promote subsequent alterations in steroid hormones levels that bind to the glucocorticoid receptor (GR) [12].
The aryl hydrocarbon receptor has roles on the metabolism of endogenous substances (mainly hormones) and exogenous substances, being a key integrator of detoxification and bioactivation of xenobiotics [66]. It has also been demonstrated that the aryl hydrocarbon receptor may affect cell proliferation, differentiation, apoptosis, and intercellular communication [67, 68], being implicated in tumor promotion upon activation by dioxins [67, 69, 70]. EDCs interaction with receptors such as AhR may compromise the normal cell functions by activating oxidative stress-sensitive signalling pathways and subsequent proinflammatory events. The coplanar PCBs, as well as other environmental contaminants are AhR agonists, establishing a link between exposure and inflammation [71].
A number of other master transcriptional regulators critical to the regulation metabolic pathways leading to obesity has also been demonstrated as possible EDC targets, including peroxisome proliferator activated receptors (PPARs), liver X receptor (LXR), farnesoid X receptor (FXR), estrogen related receptors (ERRs) and retinoid X receptors (RXRs) [42, 72]. The activation of nuclear receptors such as PPARs and LXR by EDCs interferes with the expression of an array of genes involved in lipid metabolism and inflammation [72-74]. Several other mechanisms such as oxidative stress, mitochondrial dysfunction and epigenetic modifications are possible pathways of EDCs interference in the inflammatory processes [42, 75]. Given the largely demonstrated association of inflammation and metabolic dysfunction [76], inflammatory disruption is yet another mechanism exerted by EDCs likely to favour MetS development. Since estrogens affect the immune system and inflammatory processes [77-79], the ability of some EDCs to either interfere with estrogen signalling or metabolism may contribute to the dysfunction in these metabolic and inflammatory control homeostatic mechanisms [13, 80] and actions are exemplified by changes in cytokine, adipokine and autacoid synthesis with local and/or systemic effects [81-83].
Epigenetic processes are also involved in these endocrine disrupting effects, through modifications of factors that regulate gene expression independently of modifications to the DNA sequence. These effects include changes in DNA methyltransferases, histone acetyltransferases, histone deacetylases, and histone methyltransferases with possible significant and sustained effects on gene transcription [84, 85]. Indeed, epigenetic changes mediated by dietary and environmental factors, rather than genetic changes, are a more plausible explanation for the obesity epidemic in Western countries. In this sense, the role of epigenetics is being evaluated in CVD development and progression [86]. Showing this increased interest, a recent paper demonstrated distinct disease-specific and remodelling DNA methylation signatures in nonalcoholic fatty liver disease after bariatric surgery [87]. In addition, an exciting aspect of epigenetics is the opportunity to integrate exposures as a type of ‘exposome’, leading to the expansion of environmental epigenetics field [88]. Although several EDCs have been shown to elicit modifications in DNA methylation or microRNA expression [89-91]. The knowledge regarding the epigenetic mechanisms and DNA methylation signatures related with disease progression and interaction with environmental exposures is still scarce, namely with the obesogenic effect. Additionally, one of the ultimate goals is to define novel and potentially useful disease biomarkers, namely in the interaction with environmental exposures and disease progression.
ENDOCRINE DISRUPTORS AND IN UTERO EFFECTS
Additionally, we cannot overlook the period of life when exposure has occurred and the latency from exposure to the manifestation of a disorder (a foundation for “the developmental basis of adult disease”) [92]. Perinatal exposure is an important key factor, as the developing fetus or neonate are extremely sensitive to disturbance. Indeed, the ‘thrifty-phenotype hypothesis’ suggests that the epidemiological associations between poor fetal and infant growth and the subsequent development of T2D and the MetS later in life result from the effects of poor nutrition in early life, which produces permanent changes in glucose-insulin metabolism [92, 93]. In fact, in order to promote survival, the developing organism responds to cues of environmental quality by selecting an appropriate trajectory of growth, leading to early life metabolic adaptations and preparing the organism for its likely adult environment. However, these established traits are a poor fit for the modern obesogenic environment.
An emerging additional view hypothesizes that metabolic programming of obesity and metabolic risk may be linked to perinatal exposure to EDCs at low environmentally relevant doses [94-97]. Indeed, it is well acknowledged that perinatal exposure to these compounds occurs with influence in birth weight and later health outcomes [98-103]. The hormone like activity of EDCs can disrupt the programming of endocrine signalling pathways that are established during perinatal differentiation, contributing to the development of obesity in adulthood. The specific pathways and mechanisms affected by perinatal exposure may be dependent upon the dose and the precise time of exposure, as well as other factors such as circulating endogenous hormone levels present during exposure [80].
Nevertheless, this concept needs to be supported by relevant mechanisms of action and to date, plausible molecular mechanisms include imprinting of obesity related genes or changes in cell type and number (such as adipocytes) [93, 104]. Epigenetic alterations appear to be the most likely mechanisms that could explain perinatal programming leading to later life obesity and metabolic diseases [105], giving rise to the thrifty epigenotype hypothesis. Among the several environmental exposures, bisphenol-A exposure with influence in later disease risk are becoming increasingly well documented [106, 107]. Indeed, in utero exposure to environmental relevant doses of bisphenol-A was shown to have a disruption impact on glucose homeostasis and gene expression of adult mice offspring, contributing to the development of metabolic disorders. Furthermore, the extensive harms of perinatal tobacco smoke exposure, namely as regards nicotine [108, 109], and its relationship with childhood overweight/obesity provides a proof-of-concept of how early-life exposure to EDCs can be a risk factor for obesity and MetS [109]. The mechanisms by which this perinatal exposure programs the AT and endocrine function are under investigation, but it is known that maternal tobacco smoke predisposes human and rat offspring to visceral obesity in early adulthood, possibly by programming AT dysfunction via alterations in the glucocorticoid pathway and development of leptin and insulin resistance [108, 110]. The increasing identification of genes with smoking related methylation changes in newborns and adults reinforces epigenetic modifications such as DNA methylation as one of the main mechanism by which smoking (and other EDCs) might have long lasting effects [111]. Furthermore, in animals, epigenetic effects such as DNA methylation can also be passed to successive generations, promoting a transgenerational inheritance of environmental obesogens [112, 113].
Evidently, these prenatal effects can primarily occur through changes in the main organ involved in supplying nutrients to the fetus, the placenta, affecting feto-placental development and subsequent adulthood metabolic disease. In addition to changes in placenta size and morphology, the epigenetic regulation of imprinting and transporter gene expression are also important to evaluate placental programming [114].
CONCLUDING REMARKS
Public health risks can no longer be based on the assumption that overweight and obesity are just personal choices, but rather a combination of complex events that may be contributing to the obesity epidemic and ultimately to the MetS, namely factors such as the exposure to EDCs. Recent research provides accumulating evidence to hypothesise that EDCs might have an important obesogenic and metabolic dysregulation ability that, to some extent, can also explain the interindividual variability of obesity effects in MetS development or T2D pathogenesis and probably different susceptibility to successful weight loss (Fig. 2).
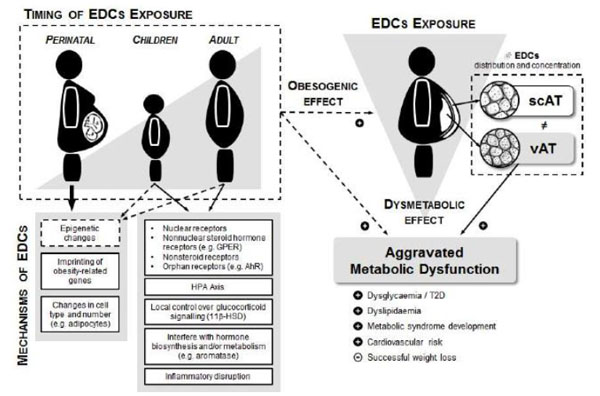
Schematic representation of the proposed mechanisms for the obesogenic and dysmetabolic effects of endocrine disrupting chemicals (EDCs), depending on timing of exposure. HPA, hypothalamic-pituitary-adrenal; T2D, type 2 diabetes; scAT, subcutaneous adipose tissue; vAT, visceral adipose tissue.
In face of these assumptions, this knowledge underline the need for public health authorities’ awareness to the extent of possible health effects triggered by these compounds. In order to act and monitor the extent of human contamination with environmental contaminants, even in countries with strict regulations. Therefore, additional research directed at understanding the nature and action of EDCs will illuminate the connection between health and the environment and may also reveal unappreciated new mechanisms regulating AT development, obesity, T2D and MetS.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

