All published articles of this journal are available on ScienceDirect.

ANP-vessel Dilator Enhances the Effect of Trametinib on Colorectal Cancer Cells via the MAPK Pathway
Authors Info & Affiliations
Abstract
Background
Colorectal cancer (CRC) is a common malignant disease that involves the interaction of both genetic and environmental factors. Trametinib (TNB) has been approved alone or in combination with dabrafenib to treat melanoma. Vessel dilator (VDL) is a cardiac hormone that possesses anticancer properties. This study evaluates TNB with VDL combined effects in CRC cells and explores the role of Mutagen-activated protein kinases (MAPK, ERK1/2) in this effect.
Methods
The HCT-15 CRC cells were treated with TNB and VDL. The MTT and ELISA assays were used to assess p-ERK, VEGF, and VEGFR levels.
Results
The results showed that combined treatment of TNB with VDL produced a significant and synergistic inhibition of HCT-15 cell growth with a combination index of less than 1. A combination treatment demonstrated no change in the expression of VEGF, inhibition of VEGFR2, and reduction of p-ERK1/2 in HCT-15 cells.
Conclusion
The findings encourage further evaluation of this combination as the combined effect is not mediated through p-ERK, VEGF, and VEGFR.
1. INTRODUCTION
Colorectal cancer (CRC) is a major public health concern worldwide. It is the third most diagnosed cancer in males and females and the second most common cause of mortality worldwide [1]. Among Jordanians, it was the second most frequent cancer in both genders and the second killer among cancers [2]. Treatment options depend on the stage of therapy, as early disease is managed surgically with adjuvant pyrimidines-based chemotherapy [3]. However, in metastatic disease, other targeted therapies may be added to conventional chemotherapy, with little or no role in surgical intervention. Targeted therapies include drugs that antagonize epidermal growth factor (EGF) or vascular endothelial growth factor (VEGF) [4]. Recently, additional genetic tests have been required to assess the involvement of specific targets, such as KRAS, NRAS, and BRAF muta tions, human epidermal receptor-2 (HER-2) expression, assessment of genetic mutations by assessing microsate llite stability, mismatch Repair proficiency, tumor muta tion burden, and Neurotrophic Tyrosine Receptor Kinase (NTRK) fusions [3].
Genetic mutations are a major cause of the malignant transformation of CRC. One of the major pathways affected is the Mitogen-Activated Protein Kinase (MAPK) pathway [5], in which RAS, RAF, and mitogen-activated extracellular kinase (MEK) are the major components. A large body of evidence shows that the activation of this pathway plays an essential role in the progression of CRC [6], which induces tumor cell proliferation, survival, orientation, and angiogenesis. This pathway is triggered by ligands, such as EGF binding to its tyrosine kinase receptor (RTK) and EGFR, activating downstream RAS, RAF, and MEK1/2. Extracellular signal-regulated kinase (ERK)1/2. Phosphory lated ERK1/2 translocates to the nucleus, activating various targets that consecutively regulate genes essential for many cellular processes, leading to proliferation, survival, invasion, invasion, and metastasis [7]. Mutations in KRAS, NRAS, and V-RAF murine sarcoma viral oncogene homolog B (BRAF) are frequently encountered in this pathway. Approximately 40% of KRAS mutations [8] and 11% of BRAF mutations have been reported in CRC [9]. The most recent NCCN guidelines include many targeted therapies for KRAS (sotorasib or Adagrasib in combination with cetuximab or panitumumab) and BRAFV600E (Encorafenib + panitumumab) mutations [3]. Mutations in MEK1/2 are less frequently encountered than RAS and RAF mutations [9]. Therefore, the latter component of this pathway is an attractive target for novel treatment approaches, as MEK is a MAPK pathway component that is downstream to both KRAS and BRAF.
Trametinib (TNB) is an orally active allosteric inhibitor of MEK1/2 [10]. This eventually inhibits cancer cell proliferation, motility, and survival [10]. The Food and Drug Administration (FDA) approved TNB for the treatment of metastatic melanoma [11], metastatic non-small cell lung cancer (NSCLC), and anaplastic thyroid cancer [12]. TNB has been investigated as a monotherapy and was found to be ineffective in patients with CRC with BRAF mutations [13] primarily because of many concomitant mechanisms of resistance, including activation of RTK and stimulation of parallel signaling pathways bypassing MEK inhibition to reactivate ERK [14]. Therefore, combining TNB with an agent that targets several signals inside the cell may improve therapeutic efficacy and enhance CRC treatment. Clinically, dabrafenib combined with TNB has been evaluated in patients with BRAF-mutant metastatic CRC (mCRC) [15] and in patients with BRAF-mutant CRC [16]. Unfor- tunately, its effectiveness is limited, unlike that in BRAF-mutant melanoma [15, 16]. In phase I/II study, TNB was investigated in combination with dabrafenib and an anti-EGFR antibody, panitumumab, in patients with BRAF V600E mCRC, and the clinical activity of the combination was confirmed [17]. Therefore, further investigation of TNB in CRC is required to achieve optimal inhibition of MAPK.
The human body synthesizes four peptide hormones within the 126-aa Atrial natriuretic peptide (ANP) prohormone that is encoded by the atrial natriuretic peptide prohormone proANP gene, long-acting natriuretic peptide (LANP), vessel dilator (VDL), kaliuretic peptide (KP) and atrial natriuretic peptide (ANP). VDL, ANP, KP, and LANP have shown anticancer effects in various types of cancer in vitro [18-23] and in vivo [24-26]. The MAPK pathway is blocked by these hormones, as they target multiple kinases in this cascade [27]. VDL, ANP, KP, and LANP exert 95%, up to 98% inhibition of the phosphorylation of MEK1/2 [25, 28-30] and up to 96% inhibition of the phosphorylation of ERK1/2 [31, 32]. Notably, those hormones did not inhibit the ERK1/2 in the normal cells [33]. The ability of those hormones to inhibit these kinases as well as inhibit DNA synthesis is mediated by intracellular messenger cyclic GMP (cGMP) inhibition [25, 28, 29, 31, 32, 34].
Vascular endothelial growth factor A (VEGF-A) promotes cancer cell growth by acting as a growth factor and stimulating the MAPK pathway [35]. These four cardiac hormones were the first dual inhibitors of VEGF and VEGFR2. VDL has the most significant anticancer properties of ANPs in many types of cancers in vitro [19, 21-23, 36] and in vivo [24-26]. In CRC, VDL treatment significantly decreased the number and proliferation of cells within 24 hrs [34]. No previous studies have investigated the combination of VDL with other agents.
In this study, we hypothesized that combining TNB and VDL would produce a significant synergistic effect on the MAPK/ERK pathway inhibition. In addition, the effect on the levels of p-ERK, VEGF, and VEGFR was assessed.
2. MATERIALS AND METHODS
In this study, the possible synergistic effect of the combination of TNB and VDL on CRC was investigated using HCT-15 cells. This study was performed using different concentrations of TNB or VDL, alone and in combination, to study their synergistic effects.
2.1. Cell Line and Materials
HCT-15 colorectal cancer cell line (ATCC® CCL-225TM) was purchased from the American Type Culture Collection (ATCC). TNB and VDL used in this study were obtained from Santa Cruz Biotechnology (Catalog # sc-364639) and Phoenix Pharmaceuticals (Catalog # 005-20), respectively. 3- [4, 5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) was obtained from Duchefa Biochemie (Cat#298-93-1), and all ELISA kits (p-ERK1/2, VEGFR2, and VEGF) were purchased from Abcam (ab176640, ab213476, ab100662, respectively).
2.2. Cell Culture
HCT-15 cells were cultured under sterile conditions in a biological safety cabinet in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The cells were incubated at 37°C with 5% CO2. For subculturing, the cells were washed with Ca2+-and Mg2+-free phosphate-buffered saline (Dulbecco's PBS W/O Calcium & Magnesium, Cat# ECB4004L) and dissociated using 0.25% EDTA-trypsin (ECB3057D) for 5-15 minutes at 37°C. Afterward, trypsin was neutralized with 10% FBS RPMI-1640 medium, and the detached cells were centrifuged, resuspended, and counted using a hemocytometer. The cells were then seeded at an appropriate number, depending on the experiment.
2.3. Drug Preparation
TNB 5 mg was stored at -20°C until use, and 200 µg of VDL was stored at 4°C until it was used. The DMSO cell culture reagent was stored at room temperature. TNB was dissolved in 0.81 ml DMSO to prepare a 10 mM stock solution, and VDL was dissolved in 1 ml sterile distilled water to prepare a 200 µg/ml (51.569 µM) stock solution. Both drugs were aliquoted and stored at -20°C.
2.4. MTT Cell Proliferation Assay
Briefly, HCT-15 cells were seeded at a density of 5×103 cells/well in a 96-well plate in 10% FBS RPMI-1640 media and allowed to attach overnight. The next day, the medium was replaced with 100 µL of fresh serum-free medium (SFM) containing various concentrations of TNB (0.39, 0.78, 1.56, 3.12, 6.24, 12.5,25 and 50 µM) and 100 µL containing various concentrations of VDL (15.6, 31.25, 62.5, 125, 250, 500, 1000, and 2000 nM) in triplicate. The control wells contained 100 µL of 0.5% DMSO and 100 µL of SFM for TNB and VDL, respectively. The cells were cultured for 48 h. Then, 10 µL of MTT solution (5 mg/ml in PBS) was added to each well, and the plates were incubated at 37°C for 2 h. The medium was then removed, and 100 µL of DMSO (solubilization solution) was added to each well to dissolve the formazan crystals. The absorbance was measured at 570 nm using a microplate reader (Biochrom EZ Read 400).
2.5. Enzyme-linked Immunosorbent Assay sample preparation
HCT-15 cells were cultured at 5×104 cells/well in 12 well-plate for 24 h. The following day, variable drug concentrations were added: 2 µM VDL, 50 µM TNB, and a combination of both and were treated vs. control. After 48 h, the medium was collected for VEGF quantification. Furthermore, to quantitate p-ERK and VEGFR2, cells were lysed by adding 250 µL/well of lysis buffer provided in the ELISA kits, followed by the collection of cell lysates for the next step.
2.6. Enzyme-linked Immunosorbent Assay VEGF, VEGFR2, and p-ERK1/2 Measurement
The quantification of VEGF, VEGFR2, and p-ERK1/2 was performed using VEGF, VEGFR2, and p-ERK1/2-ELISA kits, respectively. All measurements were performed in accordance with the manufacturer’s instructions.
2.7. Statistical Analysis
All data are presented as the mean ± SEM. Additionally, to determine significant differences between the treatment and control values, we used parametric one-way ANOVA. The significance level was set at p < 0.05. The IC50 value (the concentration that induced 50% cell growth inhibition) was obtained by applying a nonlinear regression curve fit analysis using GraphPad Prism version 9.0.0 for Windows, GraphPad Software, Boston, Massachusetts USA, www.graphpad.com.
The level of interaction between TNB and VDL was assessed using a combination index (CI), isobologram analysis, and dose reduction index.
The combination index (CI) quantitatively represents the pharmacological interactions between two compounds. A CI of 1 indicated an additive interaction, a CI of less than 1 indicated a synergistic effect, and a CI of more than 1 suggested antagonism. CI was calculated as follows [37, 38]:
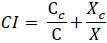 |
C and X represent the IC50 values of TNB and VDL when used alone for cell growth studies. Cc and Xc are the IC50 values of TNB and VDL, respectively, which also inhibited cell growth by 50% when used in combination.
Drug reduction index (DRI) values represent a fold decrease in the dose of individual drugs when combined compared to the dose of a single drug required to induce the same effect level. DRI values > 1 are considered favorable, allowing less toxicity while retaining the therapeutic efficacy of the individual compounds. The DRI value in this study was calculated as follows:
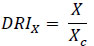 |
Where X is the IC50 value of the compound when used alone for cell growth studies, and Xc is the IC50 value for the same compound when used in combination [37, 38].
Isobologram analysis is a graphical method for evaluating the effect of equally effective concentration pairs at a single-effect level in a simple manner. It was created on a coordinate system comprising the individual drug concentrations and a straight line representing the additive effects for data points on the line was shown. The data points above the line indicate antagonistic interactions, whereas those below the line represent synergistic interactions between the two compounds when combined. A straight line in each isobologram was constructed by plotting the IC50 concentrations of TNB and VDL on the x- and y-axes, respectively. The solid straight line linking these points represents the drug concentrations for each drug that induced relatively similar growth inhibition when combined if these drugs interacted in an additive manner. The data points for each isobologram indicate the combined IC50 values of TNB and VDL. If the data point is on the line, this indicates an additive treatment effect, which lies below or above the line and represents synergistic or antagonistic effects, respectively [39].
3. RESULTS
3.1. Effect of Trametinib, Vessel Dilator, or the Combination on the Proliferation of HCT-15 Cells
Fig. (1A) shows the effect of TNB treatment on HCT-15 cell viability after 48 hrs of treatment. The results showed a dose-dependent decrease in cell viability and a significant reduction in proliferation starting at 6.25 µM. The IC50 of TNB for HCT-15 cells was 13.56 µM. Fig. (1B) shows the effect of VDL treatment on HCT-15 cell viability after 48 hrs of incubation. The results showed a dose-dependent reduction in cell viability and a significant reduction in proliferation, starting at 1000nM. The IC50 of VDL for HCT-15 cells was 1017 nM.
Fig. (1C) shows the effect of the combined treatment with TNB and VDL on the viability of HCT-15 cells after 48 hrs of incubation. However, there was no significant reduction in cell proliferation after treatment with 4 µM TNB alone. Interestingly, the combination of VDL (62.5, 125 nM) and 4 µM TNB resulted in a significant reduction in cell proliferation compared to the control.
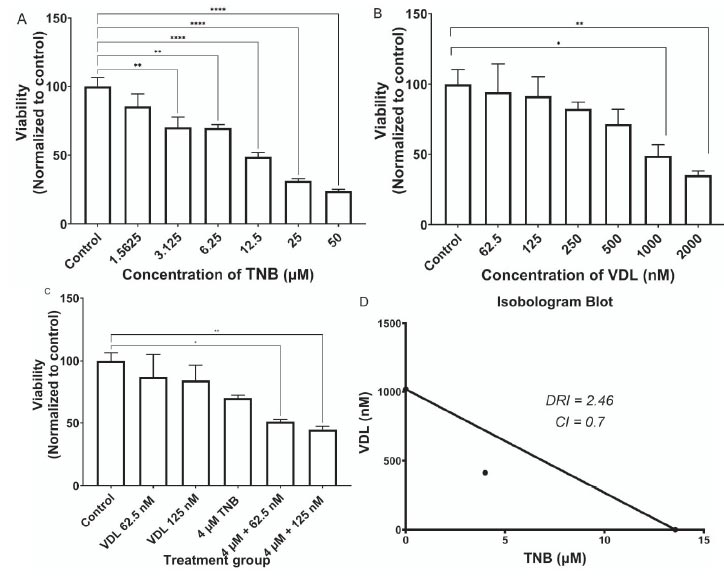
(A). Effect of TNB versus control on Proliferation of HCT-15 cells. The cell viability reduces dramatically with an increase in TNB dose. The significant effect appears at 50 μM. (* P˂ 0.05; ** P˂ 0.005;*** P˂ 0.0005;**** P˂ 0.0001) ordinary one-way ANOVA test followed by Holm-Sidak's test. (B). The effect of Vessel dilator versus control on the proliferation of HCT-15 cells. The cell viability reduces dramatically with an increase in vessel dilator dose. The significant effect appears at 2000 nM. (* P˂ 0.05; ** P˂ 0.005;*** P˂ 0.0005;**** P˂ 0.0001) ordinary one-way ANOVA test followed by Holm-Sidak's test. (C). Comparison of the effect of using 4 μM TNB separately and in combination with the different concentrations of VDL on the viability of HCT-15 cells. (*P˂0.05;** P˂ 0.005;*** P˂ 0.0005;**** P˂ 0.0001) ordinary one-way ANOVA test followed by Holm-Sidak's test. (D). Isobologram of combined antiproliferative effects of TNB and VDL in HCT-15. (D). Combination index and dose-reduction index values for combined treatment of TNB and VDL resulted in a 50% reduction in the growth of HCT-15 cells.
The isobologram analysis of the effect of the combined treatment on HCT-15 cells is shown in Fig. (1D). Isobologram indicated a synergistic interaction between TNB and VDL, as indicated by data points falling below the line of the additive effect for the combined treatments. Combination index (CI) analysis for the effect on viability of the combined treatment of TNB and VDL on HCT-15 cells is shown in Fig. (1D). The CI data agree with the isobologram analysis, in which synergistic interactions are indicated with a CI value less than 1 for combining TNB with VDL in HCT-15 cells. The dose-reduction index (DRI) also showed a multifold reduction in the growth-inhibitory dose of TNB combined with VDL (Fig. 1D).
For the next part of the experiments, we used “sub-effective” concentrations of both drugs, 2 µM for TNB and 50 nM of VDL, to study the synergistic effect of the combination.
3.2. Effect of Trametinib, Vessel Dilator, and their Combination on the Expression of VEGF, VEGFR2, and p-ERK1/2
In addition, to investigate the effects of TNB and VDL alone and in combination on the expression of VEGF, VEGFR2, and p-ERK1/2, HCT-15 cells were treated the cells with 2 µM TNB, 50 nM of VDL, or a combination of these drugs for 48 h. The results showed a significant decrease in VEGF levels with both TNB and combination treatment, with no additional effect on VDL (Fig. 2A).
Fig. (2B) shows a significant reduction in the expression of VEGFR2 in both TNB alone and in combination treatment, with no effect on VDL.
Regarding the phosphorylation of ERK1/2, the results showed a significant reduction in p-ERK1/2 expression with both TNB alone and the combination treatment, with no effect of VDL alone, as shown in Fig. (2C).
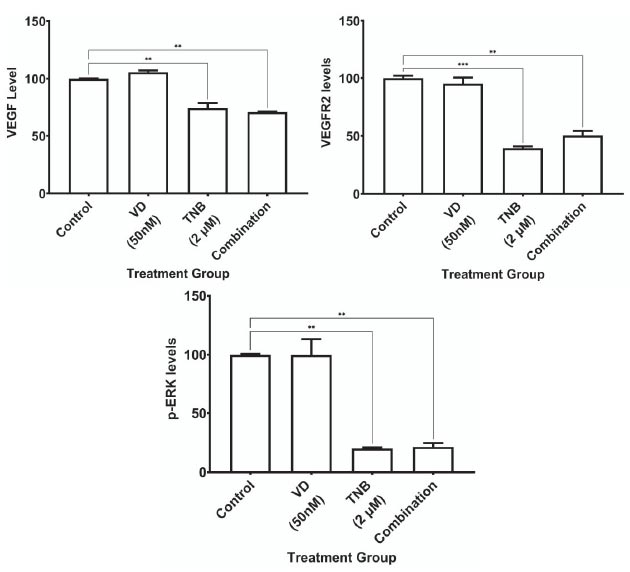
Comparison of the impact of using 2 μM TNB and 50 nM VDL separately and in combination on the expression of VEGF, VEGFR2, and p-ERK1/2. (* P˂ 0.05;** P˂ 0.005;*** P˂ 0.0005;**** P˂ 0.0001) ordinary one-way ANOVA test followed by Holm-Sidak's test.
4. DISCUSSION
The MAPK pathway plays an essential role in the progression of CRC [6]; thus, effective targeting of this pathway is a successful strategy for CRC management. Our findings showed that TNB, which inhibits MEK1/2, reduced HCT-15 cell viability in a dose-dependent manner, with a significant effect observed starting at 3.125 µM (P< 0.005). This might be expected as HCT-15 cells are RAS-mutant [40], and thus, inhibition of downstream MEK signaling will be effective in the reduction of viability of cells. In addition, VDL, which inhibits each step of the MAPK pathway, reduced the viability of HCT-15 cells in a dose-dependent manner, with a significant effect observed starting at 2 µM (P<0.05).
One group reported that TNB combined with other targeted and cytotoxic chemotherapies, especially in tumors that contain genetic alterations leading to activation of the MAPK pathway, could be more effective than monotherapy [13]. The advantages of combined treatment approaches include achieving a synergistic effect, thus allowing the use of smaller doses of each compound while maintaining a high level of effectiveness. The combination regimens of multiple drug therapies also minimize the toxicity of larger doses of single agents, overcome drug resistance, and maximize quality of life [41]. In this study, we evaluated the level of interaction between TNB and VDL treatments using CI, DRI, and isobologram. The results showed for the first time a synergistic effect of the combination on the viability of HCT-15 cells harboring a KRAS G13D activating mutation, as indicated by Ahmed, Eide [42].
Contrary to our expectations, the results of our study showed that the use of TNB, VDL, or a combination of both in HCT-15 cells did not have significant effects on VEGF, VEGFR2, or p-ERK1/2 concentrations. One study found that four cardiac hormones (VDL, LANP, KP, and ANP) inhibit the vascular endothelial growth factor (VEGF) and VEGFR2 receptor in human prostate cancer, small-cell lung cancer, and human pancreatic adenocarcinoma cells [43]. In addition, VDL can decrease VEGF by inhibiting VEGF itself or inhibiting Ras-MEK1/2-ERK1/2 kinase activity [29, 32, 44], which plays a role in the enhancement of VEGF expression [45].
Unexpectedly, in our study, VDL did not decrease the concentration of VEGF or VEGFR2 in HCT-15 cells. This study supports the findings of Hsu et al. that TNB exerts antiangiogenic activity by inhibiting VEGF expression [46]. We showed that TNB significantly inhibited the expression of both VEGF and VEGFR2 in HCT-15 cells, with no additional benefits of combination with VDL. Hsu et al. also reported that other important targets may contribute to the anti-angiogenic activity of TNB [46]. The ability of TNB to inhibit VEGFR in CRC was investigated for the first time.
In a study conducted by Sun Y and his colleagues, VDL (1 µM) was investigated on prostate cancer cells to inhibit phosphorylation of ERK1/2. Results showed that up to 96% of phosphorylation of ERK1/2 had been inhibited, and which lasted up to two hours [32]. In this study, we examined for the first time the ability of VDL to inhibit ERK1/2 phosphorylation in HCT-15 cells. The results demonstrated that VDL did not inhibit the phosphorylation of ERK 1/2 compared to the control. Similar to our previous findings, TNB blocked ERK phosphorylation, with no additional benefits for the combination.
Although combination studies using the MTT viability assay showed a synergistic effect of the combination, similar effects were not observed for VEGF and VEGFR expression or p-ERK levels. These results suggest the presence of other mechanisms that lead to increased expression of VEGF, activation of VEGFR2, and increase in p-ERK1/2, such as the PI3K/Akt, Wnt, and notch pathway [47-53]. Therefore, we postulate that blocking the ERK signaling pathway alone is not sufficient or prevents the expression of VEGF, and VEGFR2, or decreasing p-ERK levels. To this end, future gene expression studies are needed to prove this postulation using qPCR, western blotting, or IHC.
CONCLUSION
In conclusion, our study revealed that adding TNB to the vessel dilator produced a significant synergistic effect on the viability of HCT-15 cells, which was not associated with a significant decrease in ERK1/2 phosphorylation or VEGF or VEGFR2 expression. Therefore, this study encourages further investigation of the effects of this combination treatment on other signaling pathways in CRC.
AUTHOR’S CONTRIBUTION
It is hereby acknowledged that all authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| ANP | = Atrial Natriuretic Peptide |
| ATCC | = American Type Culture Collection |
| BRAF | = V-RAF Murine Sarcoma Viral Oncogene Homolog B |
| cGMP | = cyclic GMP |
| CI | = Combination Index |
| CRC | = Colorectal Cancer |
| DRI | = Drug Reduction Index |
| EGF | = Epidermal Growth Factor |
| FBS | = Fetal Bovine Serum |
| HER-2 | = Human Epidermal Receptor-2 |
| KP | = Kaliuretic Peptide |
| LANP | = Long-acting Natriuretic Peptide |
| MAPK | = Mitogen-Activated Protein Kinase |
| mCRC | = metastatic CRC |
| NTRK | = Neurotrophic Tyrosine Receptor Kinase |
| RTK | = Tyrosine Kinase Receptor |
| TNB | = Trametinib |
| VDL | = Vessel Dilator |
| VEGF | = Vascular Endothelial Growth Factor |
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available from the corresponding author [K.A] upon reasonable request.

