All published articles of this journal are available on ScienceDirect.

Molecular Characterization of Trichoderma spp. Isolates by Internal Transcribed Spacer (ITS) Region Sequencing Technique and its Use as a Biocontrol Agent
Authors Info & Affiliations
Abstract
Objective:
In the present investigation, Trichoderma spp., isolated from rhizospheric soil, has been identified by Internal Transcribed Spacer (ITS) region sequencing technique and its antagonistic activity was evaluated against A. niger.
Methods:
The sequencing analysis was done with its ITS1 region of the rRNA gene. Using the ITS1 amplified products for all isolated fungi, a bi-directional DNA sequencing was done with high quality bases (>98% - 100%). Antagonistic activity was done using dual culture technique.
Results:
All of the ITS1 nucleotide sequences obtained in this study matched 97% - 100% with the published sequence of Trichoderma spp. The results confirmed the strains as T. asperellum and T. viride with gene bank accession no. (ZTa); MK937669 and (ZTv); MK503705, respectively. When phylogenetic analysis was done for the isolates, the optimal tree with the sum of branch length = 0.69585023 and 0.10077756 for T. asperellum andT. viride, respectively, was observed. There were a total of 678 and 767 for T. asperellum and T. viride positions in the final dataset, respectively. Antagonistic activity was done for the isolated strains of Trichoderma spp. against A. niger, and it was found that T. asperellum showed maximum antagonistic activity (79.33±7.09%).
Conclusion:
The findings prolong the genome availability for relative investigations pointing out phenotypic variances to compare with Trichoderma genetic diversity. The present investigation delivered the Bases of future studies for better knowledge in understanding the complicated connections of Trichoderma spp. to be used as an effective biocontrol agent.
1. INTRODUCTION
Trichoderma spp. is a kind of fungal species that assists as an impending substitute to restrain chemicals and pathogen as resistance to crop cultivars. This aggressive and competitive property of the fungus is due to their metabolic competence [1]. Specific and precise description of Fungus is necessary for accurate diagnosis of disease and their remedy of accompanying fungal contagions. Trichoderma species are difficult to distinguish morphologically, therefore, molecular methods, including DNA sequencing and genealogical concordance phylogenetic species recognition using several unliked genes, are needed to give accurate identification of Trichoderma spp [2]. Thus, the identification of fungal species by means of traditional approaches is not as definite as by the method of genotyping. The methodology of genotyping comprises of amplification of targets that are phylogenetically informative like subunit of rRNA gene [3]. The genes encoding the rRNA are an extremely conserved sequence that is crucial for the existence of all cells. The structure of proteins and rRNA involving the ribosomes are exceedingly well-maintained during the course of evolution, as they are involved in the interaction of multifarious intermolecules and intramolecules to sustain their mechanism of protein synthesis [3-5]. Due to the antagonistic activities of Trichoderma spp., they are operative in contributing biocontrol of pathogens that are soil borne as they are simple inhabitants in the soil [6]. The chief characteristic of efficacious biological resistant strategies comprises of the manufacture, preparation, and the distribution of bioagents. It has utmost been characteristically suitable for systematic molecular investigation at the level of species, and also even within the species [7, 8]. The first attempt for phylogenetic exploration of the complete genus of Trichoderma was done by means of sequence investigation of rDNAs ITS-1 region [9]. In rDNA, an individual cluster comprises of 18S rDNA, 5.8S rDNA, 28S rDNA, External Transcribed Spacer (ETS) and Internal Transcribed Spacer 1 and 2 (ITS1 and ITS 2) [5]. For the determination of phylogenetic relationships, the spacer regions and the genes, distinctly or in grouping, are used as molecular markers [10]. However, 28S rDNA and 18S rDNA (length-conservative regions) are generally applied in the determination of phylogenetic relationships amongst groups of upper levels [10-12], although when considered at species level, regions of length variability like internal transcribed spacer 1 and 2 (ITS1 and ITS 2) are often used [13-15]. Thus, in the present study, the identification of Trichoderma spp. at species level was determined with consideration to the regions of length variability i.e Internal Transcribed Spacer (ITS).
As a biocontrol agent, Trichoderma spp works extensively virtuous (mycoparasitism mechanisms), because of its antagonistic characteristic [16, 17]. The progression ostensibly consists of Trichoderma chemotropic growth, mycoparasitic identification of the host, extra-cellular enzymes secretion, hyphae permeations, and finally, host lysis [18]. Mycoparasitism encompasses outbreak of one fungal species, directly on the other [19]. This complicated progression comprises of chronological proceedings, which comprises of identification of other fungal strain by Trichoderma spp, followed by an outbreak of cellular machinery of the host, thereafter its permeations inside the fungal host and lastly carnage the host [20]. Trichoderma spp. grows to the concerning fungal host by identifying them. Such offensive remote sensing property of Trichoderma spp. Is, to some extent, due to the consecutive formation of pathogenesis linked proteins typically chitinase and glucanase proteases [21]. Continuous secretion of exo-chitinases by Trichoderma spp. damages cell-walls of the fungal host, thereby discharging oligomers, which play a crucial part in the inhibition of fungal host [17]. Trichoderma spp. is associated with the host pathogen, loops around it and forms appressoria, thereby discharging its content [22]. Therefore it results in the formation of peptides associated with pathogenesis that services in both the entrance of Trichoderma spp. hyphae as well as cell wall content digestion [23]. The degradation of the cell of host fungus due to the formation of these biologically synthesized chemicals outcomes as parasitism. Thus, the identification of such a valuable non-pathogenic fungus would help us in commercialization and ultimately developing a superior tactic to eliminate pathogens that are soil-borne.
2. MATERIALS AND METHODS
2.1. Culture Preparation
The soil sample was collected from sugarcane cultivation field (from rhizospheric plane) located at IIM Road Lucknow, Uttar Pradesh, for isolation of Trichoderma spp. (isolation and purification processes was done by serial dilution and sub-culturing technique). Morphologically, microscopically, and biochemically identified strain was cultured and maintained on potato dextrose broth (PDB; Hi Media) and potato dextrose agar (PDA; Hi Media) plate [24]. The isolates were assigned as ZTa and ZTv for T. asperellum and T. viride, respectively. Freshly cultured pure colony was further used for DNA isolation.
2.2. Primers
The primers obtained from Bio kart India Pvt. Ltd. India, were used for Trichoderma spp. viz. ZTa and ZTv for PCR amplification were ITS 1 (´TCCGTAGGTGAACCTTGCGG) and ITS 4 (´TCCTCCGCTTATTGATATGC) with an annealing temperature of 61°C and 53°C, respectively. These primers bind to conserved regions, with corresponding positions. The PCR product was amplified that encompasses a portion of the Internal Transcribed Spacer (ITS) region. The size of the PCR product generated was varying and according to the organism tested. The same primers were used for ZTa and ZTv for direct sequencing with the same annealing temperature of 61°C and 53°C, respectively.
2.3. DNA Isolation and Sequencing
The genomic DNA of primarily identified fungal isolates was extracted and amplified using PCR, followed by the sequencing analysis, using its ITS1 region of the rRNA gene. Primarily, for genomic DNA extractions, approximately 100 mg of mycelial powder was used; mycelial powder was obtained by grinding a small portion of the fungal culture in liquid nitrogen using a mortar and pestle. Fungal genomic DNA was isolated using a modified Phenol: Chloroform method [25]. Amplification of ITS genes was carried out with the ITS 1 (TCCGTAGGTGAACCTTGCGG) and ITS 4 (TCCTCCGCTTATTGATATGC) universal primer pair, which produced ~500 bp amplicon products. PCR Master Mix (Promega ™) was used to amplify the ~500 bps region of the ITS region. A negative control (PCR mix without template DNA) was also performed in all PCR experiments. The PCR reaction conditions were set for 95°C for 2 min (1 cycle), followed by 35 cycles of denaturation at 95°C for 30s, annealing at 52°C for 30 min and extension at 72°C for 2 min, before a final extension at 72°C for 15 min (1 cycle). PCR products were subjected to purification using Omega™ PCR Purification Kit, by following the protocol of Liu et al. [26]. Purified PCR products were then subjected to an ethidium bromide-stained 1% agarose gel (Fisher Scientific) along with a 1 kb DNA ladder (Promega) to estimate the size of the amplified band. The purified PCR products were subjected to sequencing [27].
2.4. Gene Bank and BLAST
The ITS rRNA gene sequences were subjected to the BLASTn search program (National Center for Biotechnology Information) to find a similarity index between sequences. Sequences are edited using MEGA (Molecular Evolutionary Genetics Analysis) software [28]. Each sequence was subjected to an individual BLAST search to be verified in Gene Bank. The newly obtained sequences were aligned with highly similar, homologous sequences from Gene Bank using the multiple sequence alignment program MUSCLE, with default parameters. The BLASTn similarity search program was used to find homologous sequences against the NCBI nucleotide database that confirmed the species level similarity with the query sequence of the isolates.
The percentage of replicate trees, in which the associated taxa clustered together in the bootstrap test (500 replicates), were shown next to the branches [29]. This analysis involved 12 nucleotide sequences. All ambiguous positions were removed for each sequence pair (pairwise deletion option). The phylogenetic tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the maximum composite likelihood method [30, 31] and were in the units of the number of base substitutions per site. Evolutionary analyses were conducted using MEGA X [28].
2.5. Biocontrol Activity
In vitro antagonastic activity was done according to the protocol of Dennis and Webster [32]. As per the protocol, for seven days, the fungal isolates were cultured on PDA media in petri plates. The 5mm diameter disks of growing colonies of Trichoderma mycelium and Aspergillus niger were cut and transferred on other PDA media petri plates, 7 cm apart from each other. The plates were incubated for five days at 28± 1°C. A standard strain of Trichoderma spp. (MTCC167), obtained from Department of Biosciences, Integral University, was also taken and similar treatment was done. The percent inhibition of growth was determined by the method of Watanabe [33]. The experiment was done in triplicate (n=3).
3. RESULTS
3.1. Identification of Fungal Strains Using its Sequencing Technique
Morphologically, microscopically and biochemically identified strain, as reported by Haque et al. [24], were used for further identification and confirmation, using ITS sequencing molecular technique. The isolated fungal strains were identified using sequencing analysis in the present study. However, primarily, the strains were identified on the basis of morphological, microscopic, and biochemical characterization. The isolates were visually characterized on the basis of phenotypic characters like colony color (dark green and cottony whitish green colonies), growth pattern, shape and size of conidiophore, phialides and conidia were observed microscopically. The morphologically and microscopically analyzed isolates were assigned as T. asperellum (ZTa) and T. viride (ZTv), respectively. Therefore, the present sequence analysis was performed for fungal identification using its Internal Transcribed Spacer (ITS) region. The Internal Transcribed Spacer (ITS) was referred to the spacer DNA situated between the small-subunit ribosomal RNA (rRNA) and large-subunit rRNA genes in the chromosome or the corresponding transcribed region in the polycistronic rRNA precursor transcript. Using the ITS1 amplified products for all isolated fungi, a bi-directional DNA sequencing was completed with high quality (HQ) bases (>98% HQ–100% HQ). Analysis of the generated ITS1 nucleotide sequences confirmed species-identification of the fungal isolates. All of the ITS1 nucleotide sequences, obtained in this study, matched 97% - 100% with the published sequence of Trichoderma spp. with gene bank accession number of T. asperellum (ZTa), MK937669 and that of T.viride (ZTv), MK503705. No intra-specific genetic variation was noticed among the fungi. The results confirmed the Trichoderma spp. strains as T. asperellum and T. viride.
When phylogenetic analysis was done for the isolates of T. asperellum and T.viride, the following results were obtained using the Neighbor-Joining method [34]. The optimal tree with the sum of branch length = 0.69585023 and 0.10077756 for T. asperellum and T. viride, respectively, was observed. There was a total of 678 and 767 positions for T. asperellum and T. viride, respectively, in the final dataset. The phylogenetic analysis and the sequence obtained is given in Fig. (1A and B) for T. asperellum and T. viride, respectively.
3.2. Biocontrol Activity
When the antagonistic activity was tested for the isolated strains of Trichoderma spp. against A. niger, it was found that T. asperellum showed maximum antagonistic activity (79.33±7.09%) while minimum antagonistic activity was shown by the standard strain of Trichoderma spp. MTCC167 (54.33±9.50%). However, when Two-way ANOVA was applied between the three Trichoderma spp. strains, significant variation was observed with p value < 0.05 (0.024) and the effect of days on the growth were also found to be significant (p value < 0.05) (4.75E-06). The results are shown in Fig. (2).
4. DISCUSSION
The specie concept of micro-organisms (prokaryotic and eukaryotic) is well defined by Rossello´-Mora and Kampfer [35], where it was described that micro-organisms might be capable of extensive diversity range amongst the various other living organisms and can be determined by the phylogenetic analysis which shows ancestry with a common pattern. Moreover, there are various techniques for the identification of pathogenic micro-organisms for the recognition of pathogens in food crops, water, etc [36]. However, in a study by Castro-Escarpulli et al.; Bajinka and Secka; She and Bender [37-39], it was mentioned that molecular techniques were found to be the best for identification of micro-organisms, which confers precision and accuracy. The molecular methods like gene sequencing using 16S ribosomal RNA for fungal ITS sequence can be applied for the identification of organisms [40-42]. As per the previously reported findings, in the present study, we have applied the ITS sequencing technique to identify Trichoderma spp. isolates. The identification of fungus at species level by the use of ITS region (as a genetic marker) of the gene was well established [43-45]. In the past few decades, the ITS1 region sequencing technique has been extensively applied for genotyping of fungal strains, which are pathogenic to humans causing various health ailments like cutaneous infection, meningitis, allergies, and respiratory illness, etc [46]. The ITS1 region sequencing technique recognized the pathogenic human yeast and 40 species out of 106 strains have been tested [47]. It was also investigated that 44 human-pathogenic mold species were identified in 201 strains using the ITS region sequencing technique [48].
It was further reported that ITS1 region sequencing technique was used to identify and distinguish five species of Rhizopus, which can cause meningitis in human beings [49]. Infection due to Rhizopus causing intestinal mucormycosis was investigated efficaciously using the ITS region sequencing technique [50]. Since the ITS region sequencing technique is promptly being envisioned as a barcodes, suggestions have been put forward to construct a distinct set of public reference data for ITS sequences of various fungal species [51]. Efforts have been made to form a guiding principle for the genuineness and consistency of newly produced sequences of fungal ITS [52]. The query sequence of isolate T. asperellum (MK937669) showed 97.16% identity with T. asperellum (MH013956), and isolate T. viride (MK503705) showed 99.82% with T. viride of (JF304319). The findings clearly indicate that the isolated strains were confirmed as Trichoderma spp.

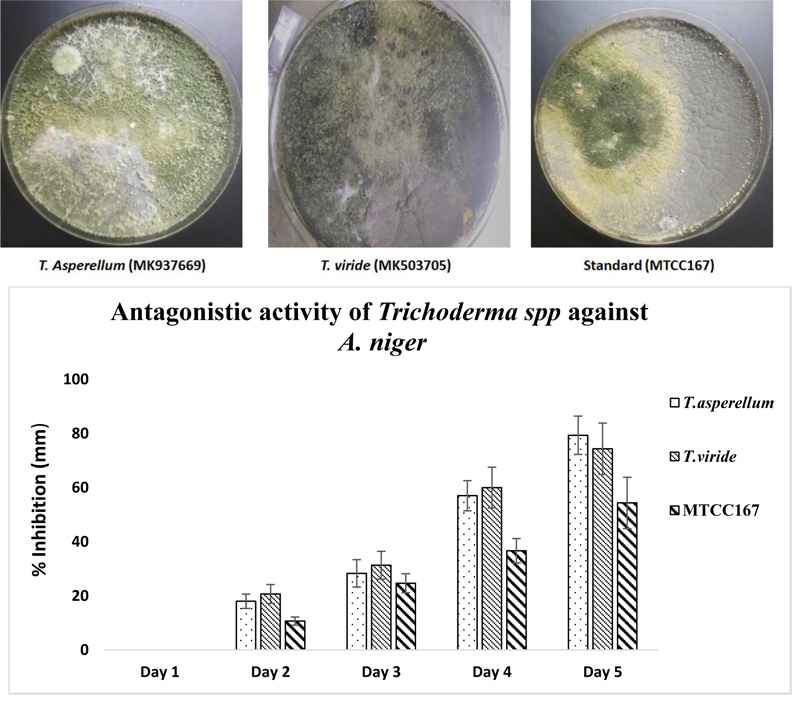
The antagonistic activity of Trichoderma spp. showed ZTa (MK937669), the better strain against A. niger. The results are in good agreement with the report of Gajera and Vakharia [53] where twelve isolates of Trichoderma spp. showed antagonistic activity against A. niger. In another study by Kurt et al. [54], Trichoderma spp. strain SJ3-4, which expresses the A. niger glucose oxidase-encoding gene, goxA, under a homologous chitinase (Nag1) promoter, increased the competence as a biocontrol agent. The mechanism of Trichoderma spp. as a biocontrol agent is well established. The synthesis of siderophores by Trichoderma spp. makes it an effective biocontrol fungus. It was elucidated that due to the synthesis of iron-chelating siderophores by Trichoderma spp., to survive against micronutrient insufficiency, makes itas an effective biocontrol agent. It has been further reported that the development of iron-chelating siderophores by Trichoderma spp. is due to the presence of other pathogenic fungi [55, 56]. Struggle for space by the pathogen from Trichoderma spp. as a biocontrol agent causes hindered root colonization, resulting in feeble establishment and poor disease cause. Thus, the observations of the present study showed that the isolated Trichoderma spp. strains are of virtuous biocontrol agents and method (ITS sequencing) [57] used for its genetic identification confirmed its occurrence.
CONCLUSION
Since fungi are very diverse at the specie level, there are several molecular techniques that have been developed for the identification of fungal species. DNA Barcoding technique is applied to a small and standardized DNA region with unique pattern and is considered as one of the most accurate and rapid method to categorize unidentified fungal species. In DNA barcoding, Internal Transcribed Spacer (ITS) region is predominantly used as it is the most sequenced region for fungal identification at species and within the species level. The ITS region is extremely polymorphic and non-coding with adequate taxonomic parts which enables to isolate sequences at species level. Molecular based techniques have endorsed more in-depth experimentation on Trichoderm, howeverthe outcomes are limited due to the lack of information of genome sequence. With this consideration, the identification of genome ZTa (MK937669) and ZTv (MK503705) is a contribution to understand the mode of action as a biocontrol agent by the use of genetic markers, which are activity-specific. The findings prolong the genome availability for relative investigations pointing to compare phenotypic variances with Trichoderma genetic diversity. It can be concluded that this investigation delivers the base for future studies for better knowledge of complicated connections of Trichoderma with multiple objectives for the improvement of effective Trichoderma strains as biocontrol agent.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available within the article.
FUNDING
None
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Authors are thankful to the Amity Institute of Biotechnology, Amity University Uttar Pradesh, Lucknow Campus and the Department of Biosciences, Integral University, Lucknow for providing necessary laboratory facilities to conduct this research.

