All published articles of this journal are available on ScienceDirect.

Actions of Estrogenic Endocrine Disrupting Chemicals on Human Prostate Stem/Progenitor Cells and Prostate Carcinogenesis
Abstract
Substantial evidences from epidemiological and animal-based studies indicate that early exposure to endocrine disrupting chemicals (EDCs) during the developmental stage results in a variety of disorders including cancer. Previous studies have demonstrated that early estrogen exposure results in life-long reprogramming of the prostate gland that leads to an increased incidence of prostatic lesions with aging. We have recently documented that bisphenol A (BPA), one of the most studied EDCs with estrogenic activity has similar effects in increasing prostate carcinogenic potential, supporting the connection between EDCs exposure and prostate cancer risk. It is well accepted that stem cells play a crucial role in development and cancer. Accumulating evidence suggest that stem cells are regulated by extrinsic factors and may be the potential target of hormonal carcinogenesis. Estrogenic EDCs which interfere with normal hormonal signaling may perturb prostate stem cell fate by directly reprogramming stem cells or breaking down the stem cell niche. Transformation of stem cells into cancer stem cells may underlie cancer initiation accounting for cancer recurrence, which becomes a critical therapeutic target of cancer management. We therefore propose that estrogenic EDCs may influence the development and progression of prostate cancer through reprogramming and transforming the prostate stem and early stage progenitor cells. In this review, we summarize our current studies and have updated recent advances highlighting estrogenic EDCs on prostate carcinogenesis by possible targeting prostate stem/progenitor cells. Using novel stem cell assays we have demonstrated that human prostate stem/progenitor cells express estrogen receptors (ER) and are directly modulated by estrogenic EDCs. Moreover, employing an in vivo humanized chimeric prostate model, we further demonstrated that estrogenic EDCs initiate and promote prostatic carcinogenesis in an androgen-supported environment. These findings support our hypothesis that prostate stem/progenitor cells may be the direct targets of estrogenic EDCs as a consequence of developmental exposure which carry permanent reprogrammed epigenetic and oncogenic events and subsequently deposit into cancer initiation and progression in adulthood.
INTRODUCTION
Prostate cancer is the most diagnosed male non-cutaneous malignancy in the Western world [1-3]. Many developed Western countries and developing Asian nations are experiencing dramatically increased incidences of this malignancy. However, the mechanism underlying this high disease incidence is not well understood. Established risk factors alone cannot provide clear clues for these unfavorable disease trends. Hormone-dependent initiation and progression of prostate cancer are the basis of hormonal treatment strategies, which have been largely focused on androgen action including androgen ablation and androgen receptor (AR) blockade [4-7]. In addition to androgens, there is compelling evidence indicating that estrogens play a central role in prostate development and carcinogenesis, although the mechanisms are not fully understood [8-11].
While prostate cancer is well known as a hormone-dependent malignancy, it is now widely accepted that other factors including age, race, genetics, diet, and environmental toxicants impact prostate cancer risk [12]. Endocrine disrupting chemicals (EDCs) are a class of environmental toxicants from naturally-occurring compounds or man-made chemicals that interfere with the endocrine system [13, 14]. In addition to their direct effects in adults, strong evidence indicates significant increased sensitivity to EDCs during the developmental stage, therefore, early-life EDC exposures predispose specific disorders and promote susceptibility to disease with aging, a phenomenon referred to the fetal basis of adult disease [15, 16]. Epidemiological and animal studies have linked early EDCs exposure to many diseases including immune system disorders, neurodevelopment defects, cardiovascular diseases, metabolic disorders and cancers [17, 18]. EDCs with estrogenic activity imitate estrogen and have been associated with prostate cancer risk [19]. These xenoestrogens are either natural compounds (e.g. phytoestrogens, inorganic arsenic and cadmium) or synthetic chemicals [e.g. bisphenol A (BPA), dioxins] that can interfere with endogenous estrogen by directly interacting with estrogen receptors (ER). This review will focus on BPA and inorganic arsenic (iAs) as examples of estrogenic EDCs.
Adult stem cells are undifferentiated cells found among differentiated cells in adult tissue that can self-renew and differentiate to repair and maintain tissue homeostasis [20, 21]. Recent advances in stem cell research indicate that stem cells may be the direct carcinogenic targets and the cells of origin in cancer initiation and progression. Malignant tumors may originate from transformation of resident normal adult stem cells into cancer stem cells, which generate abnormal progeny to initiate the tumor and continuously seed tumor growth [22-26]. Strong evidence support the existence of both normal and cancer stem cells in human prostate, although prostate stem cells are AR negative, they respond to estrogens by expressing ERs [10]. Therefore, it is highly possible that estrogenic EDCs may directly target and reprogram prostate stem cells which are subsequently transformed into cancer stem cells [27, 28]. Furthermore, the progression of prostate cancer to androgen independence and the disease relapse are increasingly being attributed to prostate cancer stem cells, which remain untouched by conventional therapies.
To directly address the above questions, we have consequently developed a method to isolate and characterize human prostate stem and progenitor cells that can regenerate normal human prostate tissue in vivo, and subsequently progress into malignancy induced by elevated estrogen in an androgen supported milieu [10, 27-29]. These in vitro prostate stem cell assays and in vivo chimeric prostate models with hormonal carcinogenic induction potential can serve as suitable models for examining stem cell perturbations and carcinogenic actions of EDCs on human prostate. We have previously demonstrated that human prostate stem and progenitor cells positively express estrogen receptors (ERα, ERβ and GPR30) and show proliferative response to both estradiol-17β (E2) and BPA [10], suggesting that E2/BPA may directly target this cell population. Indeed, we recently documented that brief developmental BPA exposure of these cells at environmentally relevant doses increases their carcinogenic susceptibility in vivo [28], similar to an estrogen effect in our previous studies in rodent prostate showing that early-life exposures to environmental estrogens increase susceptibility to prostate carcinogenesis through structural and epigenetic reprogramming [8, 30-37]. We therefore hypothesize that, early estrogenic EDCs exposure may result in epigenetic modifications in prostate stem/progenitor cells and reprogram the prostate gland altering their early development, with possible consequences for susceptibility to prostate cancer upon aging.
In this review, we will briefly summarize available evidence for estrogenic EDCs involvement in increased prostate cancer incidence; discuss recent advances in prostate stem cell research, and present evidence for reprogramming of human prostate stem/progenitor cells by estrogenic EDCs using our novel in vitro human prostate stem cell assays and in vivo chimeric prostate models.
ESTROGENIC EDCs AND PROSTATE CARCINOGENESIS
Prostate cancer is a hormone-regulated malignancy in which hormones are necessary for the growth of cancerous tissues. Despite the extensive research, the involvement of hormone in the earlier steps of carcinogenesis and the cause of prostate cancer is still unclear. Previous studies have shown that estrogens, together with androgens, play an important role in normal prostate development [38]. In rodents, estrogen exposure during fetal life can profoundly alter the developmental trajectory of the gland, sensitizing it to hyperplasia and cancer later in life [8, 33, 39, 40]. Estrogens in combination with androgens have been shown to induce prostate cancer [41, 42]. Furthermore, estrogen production in situ mediates hormonal prostatic carcinogenesis through ERα signaling [43]. More recently, chronic exposure to estrogen alone induced neoplastic transformation of rat prostate epithelial cells [44]. In men, relative serum estrogen levels increase with age as androgen levels decline. Elevated estrogens have been associated with increased prostate cancer risk [45]. Further, estrogen is locally synthesized by testosterone (T) aromatization in prostate stroma [46], while aromatase expression is induced in prostate cancer (PCa) epithelium [46] with expression elevated 30-fold in metastatic PCa compared to primary tumors [47, 48]. Recently, estrogen was shown to promote prostate cancer cells invasion through paracrine manner by inducing MMP2 in prostate stromal cells [49]. In addition, estrogen was able to regulate TMPRSS2-ERG fusion gene in patients [50] and modulate oxidative stress in prostate cancer cells [51]. More recently, dietary estrogen was reported to drive pro-carcinogenic inflammation by epigenetic gene silencing [52]. Together, these indicate an important role of estrogen in prostate cancer initiation and progression.
The upsurge in the incidence of prostate cancer in many countries has been attributed partly to the improvement in diagnostic methods and regular population screening, but this alone cannot explain the continuing rise of this disease. Studies of prostate cancer incidence changes in twins and among migrant populations show that environmental factors, including diet and chemical exposures, also contribute to this high incidence disease [53, 54]. EDCs are chemical compounds present in the environment and diet that interfere with normal hormone biosynthesis, signaling, or metabolism [55]. Today, nearly 800 environmental chemicals are identified as EDCs [17]. They are highly heterogeneous and include synthetic chemicals and natural compounds. Synthetic chemicals are industrial solvents/lubricants and their byproducts that structurally resemble natural hormones, such as polychlorinated biphenyls (PCB), polybrominated biphenyls (PBB), dioxins, plastics (e.g. BPA), plasticizers (phthalates), pesticides [methoxychlor, chlorpyrifos, dichlorodiphenyltrichloroethane (DDT)], fungicides (vinclozolin), and pharmaceutical agents [diethylstilbestrol (DES)]. Natural xenoestrogens include metals and phytoestrogens. Cadmium and inorganic arsenic are two metal or metalloid human carcinogens known to interact with ER that function as estrogenic mimics. Phytoestrogens such as genistein and coumestrol are natural chemicals found in human food. They are widely consumed and are present in infant formula [17]. A recent study reported that urinary concentrations of the phytoestrogen genistein were about 500-fold higher in infants that were fed on soy formula compared with infants they were fed on cow’s milk formula [56, 57]. This raises the concern for potential endocrine disruption by phytoestrogens.
It has been shown that dietary compounds rich in phytoestrogens can control human prostate cancer growth. Therefore, it is reasonable to evaluate and understand any potential relationship between environmental EDCs and prostate cancer risk. Evidence obtained from in vivo animal studies, in vitro human cells studies, and epidemiological studies indicate the associations between EDCs and prostate carcinogenesis and/or cancer susceptibility. The most compelling data using animal models comes from the established occupational hazards of farming, which showed increased prostate cancer rates is a function of either chronic or intermittent pesticide exposures [58-61]. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is the most toxic dioxin that causes a variety of effects including immunotoxicity, hepatotoxicity, teratogenicity, and tumor promotion [62]. In utero exposure to TCDD in mice interferes with prostate bud patterning and is associated with hyperplastic lesions in aged animals. In humans, it was shown that cadmium has a proliferative action on human prostate cells in vitro through an ER-dependent mechanism [63]. Further, epidemiologic reports have indicated a relationship between cadmium exposure and prostate cancer rates where the mechanism of carcinogenicity appears to be multifactorial [64]. Another epidemiologic study of capacitor manufacturing plant workers highly exposed to PCBs revealed a strong exposure–response relationship for prostate cancer mortality [65]. This supports previous findings of correlations between PCB 153 and 180 and prostate cancer risk in electric utility workers [66, 67]. An Agricultural Health Study is another large epidemiological study among NCI, NIEHS and EPA evaluated more than 55,000 pesticide applicators in North Carolina and Iowa since 1993 and revealed a direct link between methyl bromide exposure and increased prostate cancer rates [60]. In addition, six other common agricultural pesticides including chlorpyrifos, fonofos, coumaphos, phorate, permethrin and butylate were associated with increased prostate cancer in families with history, indicating an interaction between the environment and genetic, which further support the involvement of multiple factors that work synergistically in promoting prostate cancer [60, 68]. Specifically, chlorpyrifos, fonofos and phorate strongly inhibit two major p450s (CYP1A2 and CYP3A4) that metabolize E2, estrone and testosterone in the liver [69, 70]. A similar mechanism of endocrine disruption in vivo has been identified for PCB and polyhalogenated aromatic hydrocarbons (including dioxins, BPA and dibenzofurans) through potent inhibition of estrogen sulfotransferase which effectively elevates bioavailable estrogens in target organs [71, 72], suggesting that exposure to these compounds may disturb hormonal balance by interfering with steroid hormone metabolism which in turn contributes to increased prostate cancer risk.
EDCs are known or suspected to be capable of interfering with hormone receptors, hormone synthesis or hormone conversion. In general, there are two distinct pathways by which EDCs could disrupt hormone action: 1) a direct action on a hormone–receptor complex, for example BPA binds and activates estrogen receptors [73]; and 2) a direct action on a target protein that controls specific aspects of hormone delivery to the specific place at a specific time, such as repression of class I transcription by cadmium is mediated by the protein phosphatase 2A [74]. EDCs exhibit similar characteristics as hormones and often interfere with all processes controlled by hormones. They can exert effects on more than estrogen, androgen and thyroid hormone action. The precise mechanisms by which the chemicals induce the carcinogenic process remain to be resolved. However, in the context of current understanding of the etiology of the disease, chemicals with androgenic, anti-androgenic and estrogenic activities are likely to be relevant. Specifically, there is good evidence that organochlorine pesticides including trans-chlordane, chlordecone, and trans-nonachlor which are known to be associated with increased prostate cancer risks, have estrogen-like activities [55, 75]. Cadmium also acts as an estrogen mimic, and arsenic is capable of activating the estrogen receptors [76]. TCDD binds to the aryl hydrocarbon receptor (AhR) [62] and crosstalk between AhR and ERα is well established. Activated AhR inhibits ERa activity while ERα has a positive role in AhR signaling [77]. EDCs with estrogenic activity interfere with normal estrogen signaling, which is mediated by two estrogen receptors: ERα and ERβ [78]. Despite the low binding affinity to ERs, estrogenic EDCs are active at low concentrations which are well within the range of current human and wildlife exposures. While highlighting evidence of EDCs with estrogenic actions, we will discuss in great detail two well-known estrogenic EDCs: BPA and inorganic arsenic.
BPA and Prostate Carcinogenesis
BPA, one of the highest produced synthetic compound worldwide and an environmental pollutant with estrogenic activity, is commonly used in polycarbonate plastic containers and epoxy resin linings for many food and beverage cans, and in numerous other common household and consumer products. BPA was first synthesized in the 1890s and its estrogenic actions were identified later in 1936 [79]. BPA is so widespread in its use that there is prevalent human exposure. Although BPA has long been thought to be not persistent, recent evidence suggests longer half-lives [80]. Importantly, BPA is rapidly metabolized to BPA-glucuronide which is not hormonally active. conjugated BPA was detected in the urine of 93% of the U.S. population in a screen conducted by the Center for Disease Control [81] with significant higher levels found in infants and children [82-85]. Based on concentrations measured in food, daily dietary intakes have been estimated in Europe to be about 11 μg/kg body weight in formula-fed babies using polycarbonate plastic bottles and 1.5 μg/kg body weight in adults. Urine values may accurately reflect the actual exposure and assessment by monitoring urinary excretion of BPA metabolites. BPA has been estimated to be up to 0.16 μg/kg body weight in the US population. Although adult exposures do not necessarily imply earlier exposures in life, especially with EDCs of relatively short half-lives, there are data demonstrating nearly 5-fold higher levels of BPA in amniotic fluid compared with other body fluid, suggesting significant prenatal exposure [86].
A number of BPA effects targeting prostate in animals and human have been extensively investigated. The effects of most concern have been related to the hormonal activity of BPA targeting hormone receptors. BPA has a core structure resembling natural estrogen. We have found that prostate stem/progenitor cells express estrogen receptors and have shown response to E2 as well as BPA, indicating BPA modulation of prostate stem/progenitor population [10, 28]. Acting as a weak estrogen, BPA has a much lower affinity for the estrogen receptors ERα and ERβ than endogenous estrogen. The relative binding affinity of BPA for and its capacity to activate ER-dependent transcription is approximately 1,000 to 10,000 lower than E2 or diethylstilbestrol (DES) [87, 88]. However, BPA was capable of activating membrane ERs through non-genomic signaling pathways with an EC50 equivalent to E2, suggesting that in vivo estrogenic activity of BPA may be due to nongenomic activation of ER [89, 90]. Furthermore, BPA activates an estrogen-responsive luciferase reporter at levels that were 50% of E2 activation [91]. Thus, BPA may have a significantly lower potency than endogenous estrogens in vitro; it is still a full agonist for both ERα and ERβ. More recently, BPA has been shown to bind with high affinity to estrogen-related receptor (ERR-ã), which may be related to its ability to function as a reported endocrine disruptor [92, 93].
Prostate hyperplasia has been described after exposure to BPA [94]. Connection between BPA and prostate carcinogenic potential have also been documented [95, 96]. The prostate carcinogenic potential of BPA was recently evaluated by an expert panel convened by the EPA and the National Institute of Environmental Health Sciences [91]. Evidence from human prostate cell lines demonstrated that BPA modulate prostate cancer cell proliferation and stimulate progression for tumors with AR mutations. Using transcriptional assays, BPA was found to activate a mutated AR (AR-T877A) that is frequently found in advanced prostate cancers of patients who relapsed after androgen deprivation therapy [95]. Furthermore, BPA exposure led to unscheduled cell cycle progression and cellular proliferation in the absence of androgen in LNCaP cells that expressed this mutant AR. Because BPA had no impact on wild-type AR, these findings demonstrate that the common gain-of function AR mutant had attained the ability to utilize BPA as an agonist. Importantly, the BPA effects were greatest at lower doses of BPA compared with high-dose exposures, which represents a typical biological response of hormonally active compounds. More recently, urinary BPA levels were shown to be an independent prognostic marker in prostate cancer; exposure to BPA correlates with early-onset prostate cancer and disruption of the centrosome duplication cycle by low-dose BPA may contribute to neoplastic transformation of the prostate [97]. In addition, evidence from rodent models further supported that BPA can influence prostate carcinogenesis. In vivoanalyses of the impact of BPA on human prostate tumor growth and recurrence were performed utilizing a xenograft model [98]. At low doses equivalent to human exposures, prostate tumor size increased after BPA exposure when compared with placebo control mice. Additionally, mice in the BPA cohort demonstrated an earlier rise in prostate-specific antigen (biochemical failure), which indicates that BPA significantly shortened the time to therapeutic relapse. These outcomes underscore the need for further study of the effects of BPA on tumor progression and therapeutic efficacy. Using our rat models, we have shown that transient, neonatal low-dose BPA exposure heightened hormone-drive susceptibility to adult-onset prostate neoplastic response and precancerous lesions in aged rats [30], possibly by developmentally epigenetic reprogramming on carcinogenic risk [96, 99]. Specifically, neonatal Sprague–Dawley rats exposed to 10 ìg BPA/kg body weight during the early postnatal period (post-natal days 1, 3 and 5 when the prostate undergoes morphogenesis) significantly increased the incidence and score of adult estrogen-induced prostate intraepithelial neoplasia (PIN), the precursor lesion for prostate cancer, as compared to control rats [8, 30, 96]. These lesions in BPA-exposed rats exhibited high levels of proliferation and apoptosis suggestive of perturbed homeostasis leading to pathological lesions. This model of sensitivity to hormonal carcinogenesis is relevant to humans where relative E2 levels increase in the aging male and may contribute to prostate disease risk [100]. Furthermore, these studies identified alterations in DNA methylation patterns in multiple cell signaling genes in BPA-exposed prostates, suggest that environmentally relevant doses of BPA reprogram, the developing prostate through epigenetic alterations and subsequently result in predisposition to carcinogenesis [8, 96]. By using our in vivo humanized chimeric prostate renal graft model, We have recently demonstrated that developmental exposure to low dose BPA increases hormone-dependent cancer risk in the prostate epithelium derived from human prostate stem/progenitor cells [28]. Our ongoing studies are investigating if BPA modulates prostate stem/progenitor cells by regulating gene transcriptomes through epigenetically reprogramming of histone methylation and small non-coding RNA expression.
Arsenic and Prostate Carcinogenesis
Inorganic arsenic (iAs) is a metalloid ubiquitously distributed in nature. It is a widespread environmental contaminant and human carcinogen. Humans are thought to be exposed to arsenic primarily through drinking water and inhalation of contaminated dust from coal burning. Many countries today are still facing a potential public health crisis with arsenic-contaminated drinking water [101]. Exposure to arsenic has long been associated with a number of diseases, including non-neoplastic disorders and cancers [102-104]. In utero arsenic exposure induces carcinogenesis of skin and oncogenesis at multiple sites within the urogenital system of the adult offspring [76, 105] and whole life arsenic exposure induces lung and liver malignancies in mice [106]. It has also been shown that arsenic has transplacental carcinogenic activity in humans [107]. Fetal stem cells may be the key targets of arsenic during transplacental carcinogenesis [108].
The association between environmental inorganic arsenic exposure and prostate cancer mortality was first shown by an epidemiological study in Taiwanese men in the late 1980s [109]. In the US, increased prostate cancer incidence and mortality in certain populations was also associated with exposure to higher levels of arsenic through drinking water [110]. Several subsequent studies revealed an association between inorganic arsenic exposure and prostate cancer mortality or incidence [76, 111]. However, due to the lack of long term carcinogenic animal studies, the mechanism for the biotransformation of arsenic is not completely understood. Importantly, it has been documented that arsenic may mediate some of these effects through endocrine disruption, specifically through interaction with ER and activation of estrogen-regulated genes [112, 113]. Thus, it remains a possibility that endocrine disruption by arsenic can contribute to prostate cancer risk and further research on this topic is essential.
It has been shown that chronic iAs exposure in vitro can transform human prostate epithelial cells into a malignant phenotype toward an androgen independent state that produces aggressive carcinoma xenografts in mouse [114-116]. Recent stem cell studies showed that chronic arsenic exposure can transformed human prostate WPE-stem cells into cancer stem-like cells [117-119]. Interestingly, arsenic-transformed prostate cells can convert nearby normal stem cells into cancer stem cells [120]. Chronic arsenic exposure also caused increased cell survival, DNA damage, and increased expression of mitochondrial transcription factor A in human prostate epithelial cells [121]. Oxidative stress is another possible mechanism for arsenic action on inducing toxicity and carcinogenesis [122]. A further report indicated a role of arsenic-induced DNA methylation in oxidative DNA damage and a carcinogenic phenotype as evaluated by invasiveness, colony formation and secretion of MMPs [123]. More recently, it was showed that chronic exposure to arsenic and/or estrogen altered the expression of epigenetic regulatory genes by changing global DNA methylation and histone modification patterns in prostate epithelial cells [124]. A follow up study indicated that arsenic and estrogen exposures induced prostate epithelial cells transformation by hypermethylation-mediated silencing of MLH1 [125], a DNA mismatch repairing gene, suggesting a possible common epigenetic regulation pathway in the oncogenic event induced by arsenic and estrogen. In addition, arsenic was shown to preferentially activates the Ras/ERK pathway during malignant cell transformation in prostate epithelium, thus, it is likely to say that membrane ERs are involved. Further, several miRNAs were dysregulated in arsenic-transformed prostate epithelial cells which may account for the activation of Ras/ERK signaling, a critical step for arsenic transformation in these cells [126]. The modulation of different signaling pathways by arsenic further support that multiple mechanisms including epigenetic reprogramming are involved in the carcinogenic potential of estrogenic EDCs, which subsequently transform normal stem cells into a cancerous counterpart. Therefore, it becomes essential to further investigate the stem cell modulation by estrogenic EDCs.
HUMAN PROSTATE STEM/PROGENITOR CELLS EXPRESS ESTROGEN RECEPTORS
Primary Prostate Epithelial Cells Include Small Number of Stem/Progenitor Cells
Prostate glands are ductal structures composed of three phenotypically and functionally distinct cell types (luminal, basal, and neuroendocrine cells) embedded in a fibro-muscular stroma [127-135]. The majority of prostate epithelial cells are cuboidal exocrine luminal cells facing the ductal lumen where they express cytokeratin (CK) 8/18, NKX3.1 and AR. Luminal cells are dependent on androgens for viability and function, producing prostatic secretory proteins such as prostate specific antigen (PSA), and prostate acid phosphatase (PAP) in humans [134, 136-138]. Prostate basal epithelial cells are located adjacent to the basal lamina and are identified by expressing p63, CK5 and CK14. Neuroendocrine cells are a minor population scattered throughout the basal layer and are identified by the expression of chromogranin A and synaptophysin [139]. In addition to the three epithelial cell types, there are other rare intermediate cell types expressing prostate stem cell antigen (PSCA) and other prostate stem cell markers, representing the stem/progenitor cell population. [135, 140, 141].
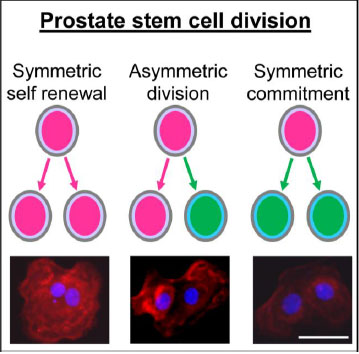
Prostate stem cell division, symmetric vs asymmetric. Immunolabeling of prostate stem cell marker CD49f in pairs of daughter cells from day 4 prostaspheres indicated 3 different types of stem cell divisions including (A) symmetric renewal, one stem cell gives rise to two identical daughter stem cells (CD49fhigh); (B) asymmetric division, one stem cell gives rise to one daughter stem cell (CD49fhigh) and one daughter progenitor cell (CD49flow); (C) symmetric commitment, one stem cell gives rise to two differentiated daughter cells (CD49flow). Prostate stem cells: in pink; prostate progenitor cells: in green; CD49f: in red. Bar=50 µm.
Adult stem cells are also called somatic stem cells with potential to replenish the cell population and repair damaged tissues to ensure proper function throughout life [21, 26]. Prostate stem cells are adult stem cells identified in both human and rodent prostate glands [141-149]. The original evidence of prostate stem cell in gland development was documented in studies using rodent models. Development of the prostate phenotype commences as the urogenital sinus (UGS) epithelial stem and progenitor cells form outgrowths or buds that penetrate into the surrounding UGS mesenchyme [127, 128, 150, 151]. Accumulated prostate stem cells with considerable growth potential have been found in the proximal region of ducts close to the UGS, and the survival of these cells does not require the presence of androgens [152, 153].
The lineage hierarchy for prostate epithelial differentiation from stem cells into differentiated basal, luminal and neuroendocrine cells has been documented in the rodent prostate and in isolated human prostate cancer stem cells [130, 135, 144, 145, 154]. Prostate stem cells are relatively growth quiescent, occasionally dividing to self-renew and generate daughter progenitor cells. [22, 147, 155, 156]. Studies across multiple systems as well as the prostate epithelium have characterized three types of stem cell divisions: 1) symmetric division, aka symmetric self-renewal, which generates two identical stem cells; 2) asymmetric division which generates a single self-renewing stem cell and a daughter cell that has entered the earliest stage of differentiation (progenitor cell); and 3) symmetric commitment division whereby a stem cell produces two daughter progenitor cells [157-159] (Fig. 1). Different types of stem cell division determine stem cell fate. Unlike the stem cell, the daughter progenitor cell has transit-amplifying capacity through rapid cell divisions. As the progenitor cell proliferative potential is exhausted, it undergoes terminal differentiation.
Isolation and Characterization of Prostate Stem/Progenitor Cells
Primary culture of prostate epithelial cells is an essential initial step in isolating prostate stem/progenitor cells from normal and diseased human prostate tissues. Fresh prostate tissue from normal organ donors and patients undergoing prostatectomy for prostate cancer or other diseases can be digested, dispersed into single cells and established as primary epithelial cell cultures according to methods characterized by Peehl and others [160]. With advances in stem cell isolation, new approaches are available to isolate stem cell populations with enhanced self-renewal capabilities from primary prostate epithelial cell cultures that contain mixed cell populations. Of these, fluorescence-activated cell sorting (FACS) and 3-D prostasphere cultures are the primary techniques employed [149, 161]. Flow cytometry is widely used for cell sorting by labeling cells with surface CD markers. Many molecules have been identified as prostate stem cell markers including Sca-1, CD133, CD44, CD117, CD49f and Trop2 [143, 147, 162-165], and the number continues to grow. While FACS is widely used for prostate stem cell research, there are several disadvantages of cell sorting including a relative low cell yield, need of multiple stem cell markers, and cell damage following labeling and sorting. In terms of functional analysis, prostate stem/progenitor cells can also be assessed using FACS side population analysis. ABCG2 is a member of the ATP binding cassette (ABC) transporters, also known as BCRP (breast cancer resistance protein), which can pump a wide variety of endogenous and exogenous compounds out of cells including Hoechst 33342 [166-168]. Widely expressed in a variety of stem cells, ABCG2 is found to be a molecular determinant of the side population phenotype and is recognized as a universal marker of stem cells [166, 167]. The Hoechst exclusion-based side population assay has proven to be a valuable technique for identifying and sorting stem and early stage progenitor cells in a variety of tissues and species. Importantly, prostate stem/progenitor cells and prostate cancer stem cells are defined by increased expression of ABCG2. The prostate stem cell side population was enriched based upon their unique ability to actively efflux Hoechst 33342. Consequently, the side-population assay can be used for the isolation and characterization of putative prostatic stem/progenitor cells [10].
Prostasphere assay is another effective approach for isolating prostate stem cells utilizing a 3-D Matrigel culture system wherein only prostate stem cells are capable of survival and self-renewal forming spheroid structures of stem and early-stage progenitor cells [10, 149, 169-171]. Using this Matrigel-slurry culture system, we are able to isolate and expand human prostate stem cells from primary human prostate epithelial cells (PrECs) in vitro. Prostate stem cells are differentially selected and increase in number with about 0.5%~2% of PrECs forming spheroid structures referred to as prostaspheres. Live video imaging was used to capture formation and size increasing of individual prostasphere. By day 4 of culture, prostate stem cells form visible prostaspheres with diameter averages of 30 mm consisting 20 to 40 cells. Prostaspheres continue to grow and increase in size to an average diameter of 80 mm. At early stages of formation, prostaspheres consist of prostate stem cells and committed progenitor cells that are actively proliferating but have not yet differentiated into cell lineages. From day 10 to 30, cells located in the spheroid center differentiate into a luminal cell phenotype (CK8/18+) with basal-type cells (p63+) in the periphery, forming double-layered prostasphere, reaching 100-150 µm in diameter (Fig. 2) [10]. Stem cell fate is determined by the type of stem cell division, either symmetric self-renewal, asymmetric division, or progenitor commitment. Several different assays have been used to monitor stem cell division. By immunostaining using prostate stem cell marker CD49f, we examined the light intensity of CD49f in paired daughter cells from day 4 prostasphere. We were able to distinguish 3 different types of cell divisions including (A) symmetric renewal, two daughter stem cells (CD49fhigh/CD49fhigh); (B) asymmetric division, one daughter stem cell (CD49fhigh) and one daughter progenitor cell (CD49flow); (C) symmetric commitment, two differentiated daughter cells (CD49flow/CD49flow) (Fig. 1). Furthermore, we have previously demonstrated that human prostate stem and progenitor cells positively express estrogen receptors (ERα, ERβ and GPR30) [10]. qPCR analysis revealed that ERβ expression increased in days 30 prostasphere as compared to days 7 prostasphere. This was accompanied by increase of prostate luminal cell markers including CK18, HOXB13, NKX3.1, AR, PSA and SOX9, and decrease of stemness gene SOX2, indicating a correlation between ERβ expression and the induction of luminal cell gene expression. Direct comparison with stem cells sorted by flow cytometry has shown that only prostate stem cells that express Trop2, CD44, and CD49f markers exhibit sphere-forming capacity in a 3-D culture system [141]. The major advantages of the prostasphere assay are the functional isolation of prostate stem/progenitor cells and the expansion capability of the stem/progenitor cell number in vitro which provides multiple research opportunities including analysis of growth and differentiation regulation.
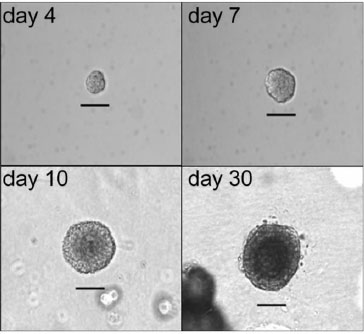
Isolation of prostate stem/progenitor cells using 3-D prostasphere culture. We isolated and expanded human prostate stem/progenitor cells from primary human prostate epithelial cells (PrECs) in vitro. Using a 3-D Matrigel culture system, prostate stem/progenitor cells were differentially selected and increased in number with about 0.5%~2% of PrECs forming spheroid structures referred to as prostaspheres. Live video imaging was used to capture formation and size increase of individual prostasphere. By day 4, prostaspheres were solid and non-canalized with diameter averages of 30-40 µm. Day 7 prostaspheres continue to grow and reach an average diameter of 50-80 µm. Day 10 prostaspheres grow to ~100 µm in size and cells located in the center start to differentiate and form double-layered structures. By day 30, prostaspheres form 100-150 µm in size and the center canalized. Bar=50 µm. (Hu et al., 2011).
In addition to stem cell self-renewal, prostate stem cells can differentiate under certain condition. The dysregulation of stem cell differentiation could be one of the early events of cell transformation and carcinogenesis. Studying the effects of EDCs on prostate stem cell differentiation would be a useful model for evaluating their carcinogenic potential.
In our previous studies, we have reported several different strategies to achieve cell differentiation of prostate stem/progenitor cells. We grew cultures of prostasphere for up to 30 days with co-culture of stromal cells and treatment with differentiating factors such as 10 nM dihydrotestosterone (DHT) or 25 ng/ml hepatocyte growth factor (HGF) [10, 29, 140]. The differentiation of prostasphere cells is characterized by the formation of ductal branches as well as lumen-like structures, thus recapitulating events in early prostate growth and differentiation [10]. Differentiation of prostate stem cells is also indicated by increased expression of luminal markers including CK8/18, NKX3.1, AR and PSA as well as basal cell markers CK5 and p63. Recently, we have also found that all-trans retinoic acid (100 nM) can directly drive prostate stem/progenitor cells into a differentiation phenotype as indicated by the early formation of a double layered prostasphere at day 7 as well as the upregulation of CK18 and HOXB13 gene expression representing luminal epithelial cells differentiation [29]. We anticipate that EDCs may interfere with retinoid signaling that directly alter human prostate stem cell differentiation capacity with potentially detrimental outcomes with regards to prostate carcinogenesis.
ACTION OF ESTROGENIC EDCS ON HUMAN PROSTATE STEM/PROGENITOR CELLS
Accumulating evidences support a central role for estrogens in prostate cancer. The E2:T ratio rises with age in men and is associated with increased prostate cancer incidence [172]. Additionally, the estrogen metabolizing gene polymorphisms and elevated urine hydroxy-estrone ratios are associated with a higher prostate cancer risk [173-175]. Aromatase expression is increased in prostate cancers upon progression to metastatic disease [89]. Marked alterations in ER expression with cancer progression [176, 177] further support the involvement of estrogens in the etiology and progression of this disease. Furthermore, tissue recombinant experiments using pre-initiated, human prostate BPH-1 cells have shown that elevated E2 and T were able to promote these cells into invasive cancers [42, 178]. Multiple studies over the past several decades using animal models have provided strong evidence of a carcinogenic role for estrogens in the prostate [90, 179-181]. However, there is no direct evidence that estrogens initiate prostate cancer in humans. The role of prostate stem cells in hormonal carcinogenesis had not been clarified.
The cell(s) of origin for prostate cancer may include luminal, basal, neuroendocrine, stem and progenitor cells [24, 182-184], while normal adult stem cells are involved in tissue maintenance and repair throughout life, increasing evidence indicate that prostate cancers contain cancer stem cells that continuously seed and maintain tumor growth [21, 23, 25, 185, 186]. Traditional convention therapies eradicate the majority of cancer cells within a prostate tumor, most patients with advanced cancer eventually progress to androgen-independent, metastatic disease that remains essentially incurable by current treatment strategies. Recent evidence has shown that cancer stem cells are a subset of tumor cells that appear to be therapy-resistant and are responsible for maintaining cancer growth which may be the underlying cause of disease relapse [22, 146, 149, 187-189]. Thus, understanding the regulation of both normal stem cells and cancer stem cells may provide new insight into the origin and treatment of prostate cancer. Towards this end, identification and characterization of these rare cell populations has been a major research effort during the past decade with marked progress utilizing flow cytometry and prostasphere cultures [141, 162, 190]. Compared to differentiated cells, stem cells stay relative quiescence allowing for a longer life span and are more susceptible to environmental injuries over time and having capacity to transmit their “injury memory” to the differentiated progeny [191]. Since the prostate gland is most susceptible to environmental insults during early development, it is reasonable to predict that prostate stem and early stage progenitor cells may be the primary targets of estrogenic exposures throughout life [10]. Herein, we have employed our established in vitro and in vivo prostate stem cell assays to test whether prostate stem cells are the potential targets of estrogenic EDCs, and to evaluate whether early life exposure to estrogenic EDCs can actually increase the susceptibility to prostate cancer.
We have documented that although negative for AR mRNA and protein, normal human prostate stem/progenitorcells express robust levels of all known ERs, including ERα, ERβ, and GPR30, and show proliferative response to E2, suggesting that E2 may directly target this cell population [10]. Prostate stem cells can undergo self-renewal or differentiation. Stem cell fate is determined by stem cell division, either symmetric or asymmetric division. Hormones including E2 and estrogenic EDCs may modulate prostate stem cell self-renewal and differentiation through regulation of stem cell division, by interfering the balance of stem cell homeostasis (Fig. 3A). Immortal strand DNA hypothesis proposed by Cairns [192, 193] suggests when stem cells undergo asymmetric division, parental DNA from mother stem cells segregate into one daughter stem cell and the daughter cell acquires newly synthesized DNA becomes a committed progenitor cell. Using BrdU labeling assays we were able to track prostate stem cell division in prostasphere cells. When prostate stem cells undergo asymmetric division, one daughter cell retains BrdU that keeps the stemness phenotype, representing the daughter prostate stem cell. The other daughter cell begins to enter early differentiation and express differentiation marker CK14, which is then a committed prostate basal progenitor cell (Fig. 3B). This novel assay will allow us to directly examine the estrogenic EDCs effect on prostate stem cell division. Using in vitro prostate stem cell assays, we further tested several estrogenic EDCs to determine if they may similarly be capable of affecting prostate stem cell self-renewal and/or differentiation capabilities. Interestingly, BPA treatment at 100 and 1000 nM significantly increased the percentage of prostate stem/progenitor cells side population in primary prostate epithelial cells in 3-D prostasphere culture. BPA at 10 nM significantly increased prostasphere number similar to the E2 exposures [28]. Although arsenic at a dose of 5 µM inhibited prostasphere formation and decreased prostate stem cell number (Fig. 4A), it stimulated Akt activity in prostasphere cells by increasing pAkt/total Akt ratios, indicating early event of cell transformation (Fig. 4B). Together, these results demonstrate that normal human prostate stem/progenitor cells are responsive to estrogenic EDCs, implicating these cells as direct estrogen targets. These preliminary data suggest that estrogenic EDCs with estrogenic-type actions may target and affect prostate stem/progenitor cells self-renewal and initiate cell transformation perhaps mediated through divergent ERs and other molecular signaling pathways. As stem cell alterations are long lasting, such perturbations may contribute to prostate cancer risk throughout life.
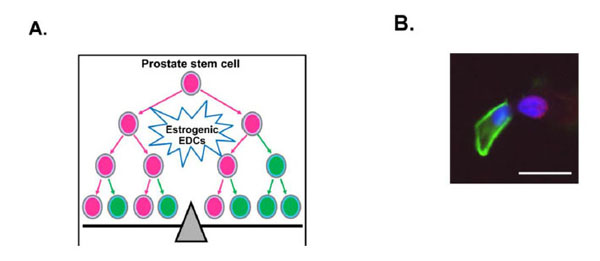
Possible EDCs effect on prostate stem cell division and its observation by BrdU labeling. (A) Stem cell fate, self-renewal or differentiation, is determined by stem cell division. Estrogenic EDCs may regulate prostate stem cell fate through modulation of prostate stem cell division, either symmetrically or asymmetrically, which will directly interrupt the balance of stem cell pool and tissue homeostasis. Prostate stem cells: pink; prostate progenitor cells: green. (B) PrEC cells were cultured for 6 days in the presence of 1 µM BrdU. BrdU labeled PrEC cells were plated into 3-D prostasphere culture. Day 4 prostaspheres were dispersed into single cells and grew on chamber slides overnight to allow cell division. Double immunostaining was performed using BrdU and CK14 antibodies. Prostate stem cells from day 4 prostaspheres undergo three different types of cell division as described in Fig. (2). A representative asymmetric stem cell division is shown. In two paired daughter cells, one cell retaining BrdU (red) represents prostate stem cell; the other cell expressing CK14 (green) is a committed basal progenitor cell. Bar=50 µm.
Today, more than ten animal models for prostate carcinogenesis have been described. Evidences from rodent model have shown that early life exposure to EDCs increase prostate cancer susceptibility [19, 32, 96, 194]. However, none of these models used human prostate cells starting with the normal disease free stage. In human, studies are primarily limited to epidemiology studies and studies using human cancer cell lines [33, 76, 97, 195]. Direct connection of EDCs with human prostate cancer risk in vivo would be ideally studied in a model by using normal human cells in animals. The regeneration ability in vivo is another important feature of prostate stem/progenitor cells that can give rise to ductal structures containing both basal and luminal cells. To directly test the carcinogenic potential of estrogenic EDCs, by using the isolated human prostate stem and progenitor cells, we have been able to regenerate a chimeric normal human prostate tissue in vivo and subsequently drive the carcinogenesis progression induced by elevated estrogen in an androgen supported milieu [10]. Formation of human-like prostate tissue was achieved by mixing human prostate stem/progenitor cells with rat embryonic urogenital sinus mesenchyme and grafted under the kidney capsule of nude mice. Normal glandular structures that produce prostate specific antigen (PSA) formed within one month. Recent evidence showed that transformation of prostate stem cells is sufficient for prostate cancer initiation in rodent and human models [146, 183, 196]. Therefore, we further treated the host mice bearing the human-like prostate structures with testosterone and estradiol-17β (T+E) to induce hormonal carcinogenesis. Prostate epithelial pathology were observed over a 1-4 month period, progressing from hyperplasia to prostatic intraepithelial neoplasia (PIN) and adenocarcinoma at a relatively low incidence [10]. This is the first model showing evidence of human prostate stem/progenitor cells as direct estrogen targets and that estrogen, in an androgen-supported milieu, is a carcinogen for human prostate epithelium. The relatively low cancer incidence in this novel system will allow the direct assessment of whether early EDCs exposure are capable of increasing susceptibility of estrogen-induced PIN lesions and prostate cancer in human prostate tissue. Indeed, using this novel humanized model, our recent studies identify for the first time that developmental in vivo exposure of the human prostatic epithelium to low-doses of BPA significantly increases the susceptibility of the human prostate epithelium to hormonal carcinogenesis [28]. Taken together, our in vivo data further supports the possibility that human prostate stem and early progenitor cell populations might be direct targets of estrogenic EDCs increasing the susceptibility to hormone-induced carcinogenesis in aging males.
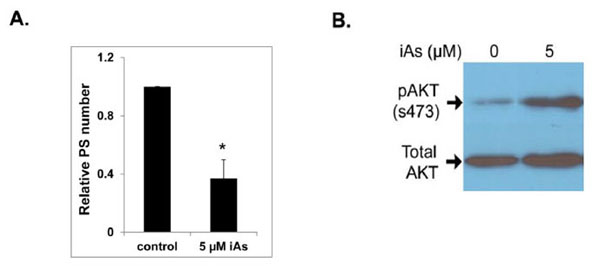
Effects of 5 µM sodium arsenite (iAs) on prostasphere formation and AKT phosphorylation. (A) PrEC cells were cultured in 3-D to form prostaspheres for 7 days in the absence or presence of 5 µM iAs. Numbers of prostaspheres (size >40 µm) were counted. iAs treatment significantly decreased the prostasphere number (N=4, P<0.05). (B) iAs treated prostaspheres were harvested for protein isolation and levels of pAKT/total AKT were evaluated by western blot analysis. iAs treatment greatly increased pAKT levels as compared to vehicle. Experiments were repeated three times. A representative blot is shown.
CONCLUSION
In summary, stem cell fate is critical in maintaining the balance of tissue homeostasis. In addition to their self-renewal ability, normal prostate stem cells are responsible for the replacement of differentiated prostate epithelial cells within continuously regenerating prostate tissue while terminal differentiated prostate epithelial cells undergo apoptosis. Exposure to EDCs can actually modulate prostate stem cell self-renewal, affect stem cell differentiation and cell apoptosis, as well as drive cell transformation. EDCs may regulate prostate stem cell fate through reprogramming the stem cell division, either symmetrically or asymmetrically (Fig. 5). Our ongoing studies are underway to address this specific question. The new information from our in vitro and in vivo models will gain great interest and will be highly valuable to the field and regulatory communities in terms of presenting strong and compelling evidence for the adverse impact of estrogenic EDCs on humans. These models can contribute to long-term growth in the research enterprise on endocrine disruptors and can serve as a basis for studying carcinogenic effects of estrogenic EDCs. Moreover, it is highly possible that estrogenic EDCs acting through the ER signaling pathway may directly reprogram or transform prostate stem/progenitor cells and subsequently promote tumor initiating capacity. Estrogenic EDCs were recently shown to reprogram organ development by modifying epigenetic mechanisms [96, 197]. Future studies will investigate multiple epigenetic modifications in the human prostate stem/progenitor cells by estrogenic EDCs. Finally, through the novel humanized model systems employing prostate stem/progenitor cells as described, we have potentially provided direct evidences connecting early-life estrogenic EDCs exposures and human prostate carcinogenesis.
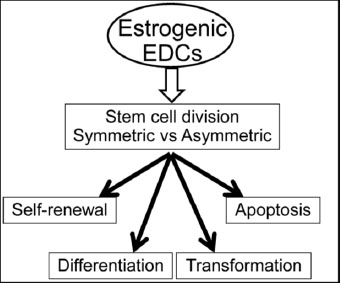
The maintenance of tissue homeostasis balance is determined by stem cell fate, including stem cell self-renewal, tissue regeneration and repair, differentiation potential and apoptosis. EDCs are able to modulate prostate stem cell self-renewal, affect stem cell differentiation and cell apoptosis, as well as drive cell transformation and subsequently initiate tumorigenesis. Stem cell division, either symmetrically or asymmetrically, is critical in determining stem cell fate. EDCs may regulate prostate stem cell fate through reprogramming the stem cell division, either symmetric or asymmetric.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
Acknowledgements
This work was supported by NIH grants RC2 ES018758, RO1ES022071 and RO1CA172220.

